- Home
- News
- Spotlight on Science
- Enzyme waiting room...
Enzyme waiting room controls carbon dioxide reduction
18-05-2026
Some enzymes are remarkably good at reducing CO₂, but what makes them so efficient? By pressurising enzyme crystals with gas at the ESRF’s High-Pressure Freezing Laboratory and collecting X-ray data at the macromolecular crystallography beamlines, researchers have mapped the routes CO₂ takes as it travels through the protein, revealing a 'waiting room' where substrate molecules queue up just outside the catalytic centre before being processed. Understanding this internal traffic system opens a path toward engineering faster, more effective enzymes for carbon capture.
Share
The challenge
Climate change remains one of the most urgent global challenges facing humanity. The past decade includes the 10 warmest years on record, and the global average temperature is now approximately 1.3°C above pre-industrial levels. This warming is primarily driven by rising greenhouse gas emissions, particularly CO2. Addressing this issue requires the development of decarbonised energy technologies and efficient strategies to capture and convert CO2. However, CO2 is a very stable molecule, making its industrial conversion into useful products scientifically and technologically challenging.
Formate dehydrogenases are highly efficient natural enzymes capable of reducing CO2 under mild conditions, yet their catalytic mechanisms remain incompletely understood. A deeper understanding of these processes is essential to design improved enzymes for industrial applications. In particular, how CO2 and formate access, bind, and react at the active site are key unresolved questions as these processes involve transient molecular events that are difficult to capture experimentally.
Using the ESRF’s macromolecular crystallography (MX) beamlines for high-resolution crystallography, combined with gas-pressurised crystals using the High-Pressure Freezing Laboratory (HPMX) sample preparation facility [1], researchers have been able to map in detail how gas molecules bind and transport in a formate dehydrogenase enzyme.
The experiment
High-pressure gas derivatisation was used to investigate how substrates diffuse from the protein surface to the buried active site of Nitratidesulfovibrio vulgaris formate dehydrogenase (FdhAB). Crystals were soaked and flash-cooled under high-pressure in the presence of CO2, O2, and Kr gases. The resulting crystal structures, determined by X-ray diffraction at MX beamlines ID23-1, ID23-2, ID30A-3, and ID30B, revealed multiple gas-binding sites within the enzyme (Figures 1 and 2).
Click image to enlarge
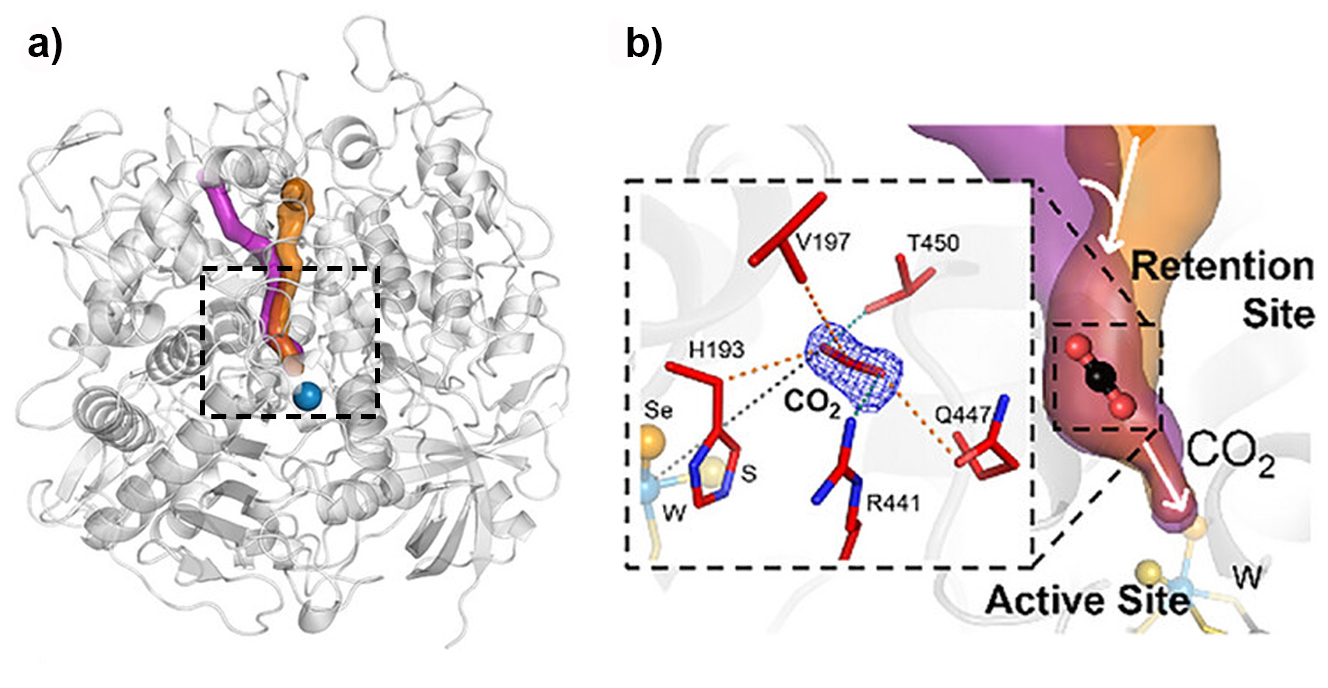
Fig. 1: The retention site identified in Nitratidesulfovibrio vulgaris formate dehydrogenase (FdhAB).
a) The main access tunnel (orange), which includes the retention site, and a CO2-specific branch (violet) are shown. b) The zoomed view highlights the CO2 molecule bound within the retention site and the surrounding residues (sticks). The tungsten (W) active site is shown below, with the W ion, sulfido group, and SeCys192 selenium atom represented as spheres (light blue, yellow, and orange, respectively).
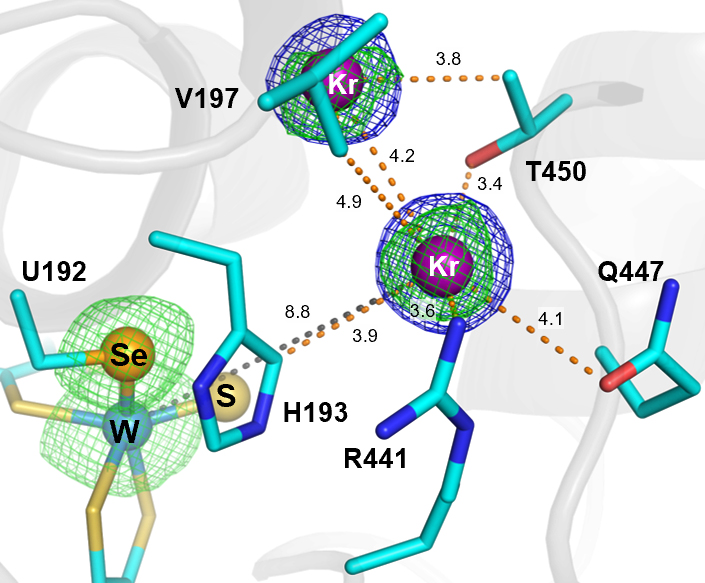
Comparison with additional FdhAB structures containing other ligands, including formate, formamide, and glycerol, identified a conserved region within the main access tunnel that acts as a substrate retention site (‘waiting room’). This site, located close to the catalytic centre, is consistently occupied by different ligands across all the crystal structures determined in this study. Molecular dynamics simulations and analysis of key residue variants lining this region support its functional importance in substrate handling and CO2 reduction within the enzyme.
Click image to enlarge
Fig. 2: Close-up of the retention site showing krypton atoms within the main access tunnel to the tungsten (W) active site, including one located at the retention site. Electron density maps are shown as blue (2Fo–Fc, 1σ) and green (anomalous difference, 3σ) meshes. The W ion, sulfido group, and SeCys192 selenium atom are represented as spheres (light blue, yellow, and orange, respectively). Ligand–W distances and electrostatic interactions are indicated by dashed lines (grey and orange, respectively), with distances given in Å.
The impact
These findings provide new insights into the functioning of metal-dependent formate dehydrogenases, addressing key questions about CO2 conversion under mild conditions [2]. The identification of a retention site reveals the molecular determinants underlying the enzyme’s high activity and selectivity. This understanding offers a basis for the rational design of improved enzymes for CO2 reduction in industrial applications. More broadly, the results contribute to the development of more effective catalysts for CO2 conversion.
|
Principal publication
|
About the beamlines
The ESRF’s Macromolecular X-ray Crystallography, Imaging & Scattering (MXIS) group operates a world-leading suite of beamlines and support facilities dedicated to the study of biological macromolecules and soft matter. It comprises five macromolecular crystallography beamlines: two tuneable (ID23-1 and ID30B), a fully automated “hands-free” (ID30A-1), and two microfocus/minibeam beamlines (ID23-2 and ID30A-3); together with a serial crystallography beamline for time-resolved and room-temperature structure determination (ID29); two solution scattering beamlines (BM29 and ID02) covering bioSAXS and time-resolved (U)SAXS/WAXS; and a cryo-EM microscope (CM01) for single-particle analysis and cryo-electron tomography.
Complementary support infrastructure includes the High Pressure Freezing Laboratory for Macromolecular Crystallography (HPMX), in-crystallo spectroscopy (icOS), a dedicated biology laboratory (CIBB) and biomedical facility (BMF) for sample preparation and cryo-preservation, alongside biophysical characterisation through the Partnership for Soft Condensed Matter (PSCM). For more information, please click here.
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.