- Home
- News
- Spotlight on Science
- Time-resolved Hard...
Time-resolved Hard X-ray diffraction (HXRD) with integral infrared spectroscopy: a new experiment to illuminate fundamental properties of reactive nanoparticulate materials
29-06-2010
A novel combination of infrared spectroscopy and X-ray diffraction, using very hard X-rays, illuminates the structural reactive behaviour of working catalysts. It reveals previously hidden aspects of the fundamental interactions occurring between small molecules and noble metal nanoparticles.
Share
Many functional materials, including heterogeneous catalysts, exhibit structural variation on a wide range of length scales - from the chemical bond, through nanosized component phases, to the micrometre scale. Whilst a catalytic event is intrinsically a molecular level process, efficient long term catalytic function often relies upon structuring and synergies that occur on a wide range of length scales: from the ubiquitous “nano”, to the microscale [1]. This layered complexity explains the great interest of these materials, but has a consequence that no single method can address all the required structural-reactive facets.
By taking advantage of the intrinsic properties of very hard X-rays, that greatly reduce restrictions due to sample environments required for in-operando studies of working materials, we have demonstrated a novel, time-resolving combination of hard X-ray diffraction (HXRD) and diffuse reflectance infrared spectroscopy (DRIFTS). This allows us to specify exactly how Al2O3 supported Pd (ca. 3 nm diameter) nanoparticles actively participate in some prototypical chemistry that lies at the very heart of car catalyst function, i.e. the efficient conversion of CO to CO2 and the reduction of NOx to N2.
According to Bragg’s law (λ/d = 2sinθ), diffraction is compressed into smaller solid angles as the X-ray energy is increased. Consequently, at 86.8 keV (λ = 0.143 Å) we can collect a sufficiently large Q(Å-1) range for a viable diffraction experiment using a sample environment that was previously developed for combined EXAFS and DRIFTS [2]. The sample environment provides a cone with an opening of ±10° for X-ray transmission. By coupling it to a flat panel Si detector (Pixium) [3], and measuring at an undulator based hard X-ray source(ID15), a new experiment was devised that allows reactive processes to be studied as they happen, from both local order (EXAFS) and long range order (HXRD) perspectives, whilst retaining infrared spectroscopy as a common probe of molecular speciation.
Intrinsic to car catalyst operation is a rapid alternation between reducing and oxidising environments. This experiment aims to model some of the aspects of the actual operation of catalysts of this type to establish how Pd nanoparticles respond to such changing conditions and how this might relate to the chemistry that they facilitate (Figure 1).
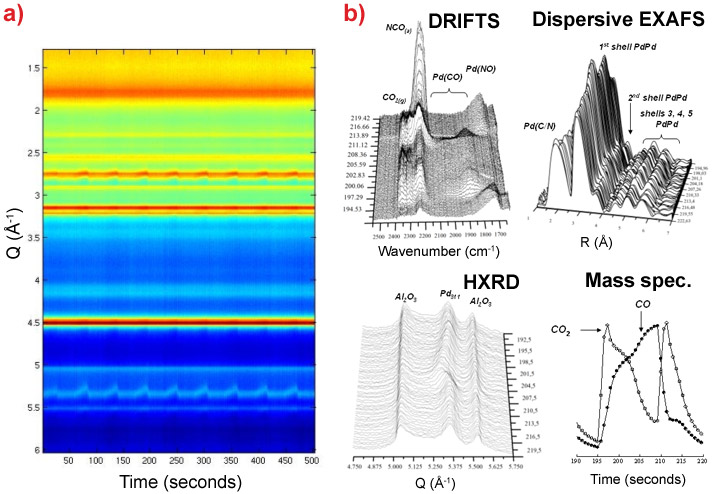 |
|
Figure 1. a) Colour map representation of the time dependent powder diffraction of supported Pd nanoparticles during CO/NO cycling at 673 K (10 cycles). b) Data from the four probes –DRIFTS, EXAFS, MS and HXRD ([311] reflection from Pd, straddled by 2 Al2O3 reflections) that may be applied in situ using the new experiment and pertaining to a single NO-CO-NO cycle. Each of the X-ray techniques may be used in conjunction with simultaneous time resolved DRIFTS and MS. |
In HXRD we see a superposition of diffraction features arising from the Al2O3 support and the Pd nanoparticles (as indicated). Whilst the features due to Al2O3 remain constant during the reactive chemistry, those originating from the Pd nanoparticles do not. This is most clearly observed in the case of the [311] reflection and indicates that the lattice constant of the Pd nanoparticles slowly increases during the CO cycle. In NO/He this is rapidly reversed and a “sawtooth” behaviour is apparent in the peak position, which is shown in Figure 1a. Whilst clearly visible in the HXRD pattern, it is not immediately evident from either the EXAFS spectra or the infrared spectra that such a process is occurring.
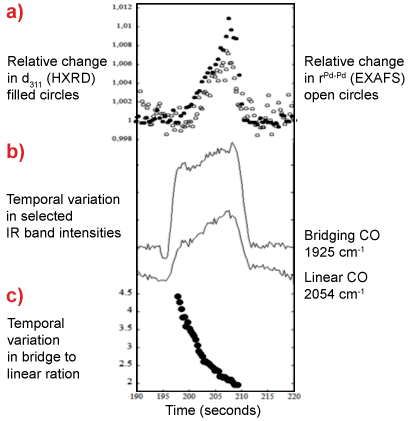 |
|
Figure 2. Temporal behaviour of 2wt%Pd/Al2O3 during the NO/CO/NO cycle at 673 K: a) relative change in first shell Pd-Pd distance (EXAFS) and d spacing of the Pd[311] reflection (HXRD); b) intensity of bridging and linear CO bands (DRIFTS); c) bridge CO to linear CO ratio (DRIFTS) during the CO cycle. |
Figure 2 shows the relationships between the partitioning of adsorbed CO in bridged and linear sites to the expansion of the Pd lattice during CO exposure. The results lead to the conclusion that some of the CO molecules adsorbed on the Pd dissociate and a net disproportionation (the Boudouard reaction) results i.e.
2CO → 1CO2 + C [1]
The online mass spectroscopy (MS) confirms the formation of CO2 during the CO exposure but cannot reveal its origin or what happens to the atomic carbon left behind. The HXRD, being sensitive to the bulk structure of the Pd nanoparticles, shows that this remaining atomic carbon is transiently stored within the particles themselves: this causes the Pd lattice to expand in the manner observed by HXRD. The progression of this expansion, when compared to the information extracted from the DRIFTS, indicates that this process starts with a rapidly formed CO adlayer. Moreover, by combining the HXRD directly with the DRIFTS, we can show that this change in structure has a significant consequence for the partitioning of the remaining CO adsorbed at the surface: occupation of linearly adsorbed CO sites is significantly promoted.
In summary, we have demonstrated a new experiment to study the reactive processes that occur in heterogeneous catalysis, solid state chemistry, and numerous other energy-related arenas that rely upon the ability of materials to dynamically store and release gases according to the process conditions in use. In the current example we have directly shown that the supported Pd nanoparticles actively participate in this chemistry by storing and eventually releasing atomic carbon when the reactive environment returns to a net oxidising state.
References
[1] See for example, B.M. Weckhuysen, Angewandte Chemie-International Edition 48, 4910-4943 (2009).
[2] M.A. Newton, Topics in Catalysis 52, 1410-1424 (2009).
[3] J.E. Daniels, M. Drakopoulos, J. Synchtrotron Rad. 16, 463 (2009).
Principal publication and authors
Combining time resolved hard X-ray diffraction (XRD) and diffuse reflectance infrared spectroscopy (DRIFTS) to illuminate CO dissociation and transient carbon storage by supported Pd nanoparticles during CO/NO cycling, M.A. Newton,(a) M. Di Michiel, (a) A. Kubacka, (b), M. Fernandez-Garcia (b), J. Am. Chem. Soc. 132, 4540 (2010); Multitechnique analysis of supported Pd particles upon dynamic, cycling CO/NO conditions: size-dependence of the structure-activity relationship, A. Kubacka (b), A. Martínez-Arias (b), M. Fernández-García (b), M. Di Michiel (a), M.A. Newton (a), J. Catal. 270, 275 (2010).
(a) ESRF
(b) Instituto de Catálisis y Petroleoqíimica, CSIC, Madrid (Spain)
Top image: Periodic structural change in ~3 nm diameter Pd nanoparticles during model catalysis for emission control observed in situ.
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.