- Home
- News
- Spotlight on Science
- Ultrafast laser...
Ultrafast laser switching in a photochromic film visualised by time-resolved X-ray diffraction
09-02-2010
A [2+2] photo-cycloaddition reaction in an organic crystal film was triggered by a fast laser pulse and studied with X-ray diffraction. This reaction and its thermally-activated reverse reaction were followed on the picosecond timescale, and intricate details of the reaction kinetics were revealed.
Share
Photoinduced processes in organic crystals [1] and their potential for applications in a variety of photosensitive devices [2] has made them the focus of much research not only in optical data storage [3] but also in template-controlled synthesis in which one of the reaction steps is photo-induced. Solid-state photochemical reactions are highly dependent on the geometry of the reacting compounds and their products. Much experimental work has been undertaken to understand the thermodynamical properties of solids undergoing solid-state reactions [4], the influence of the spatial arrangements of the molecules within the lattice, optimisation of reaction conditions (i.e., type of solvent for dispersal of crystals), and temperature dependence. However, very little is known about the kinetics of such reactions. Time-resolved crystallography permits the structural changes in such solid state reactions to be followed as a function of time, revealing the reaction mechanism and its dependency on external parameters. Photoinduced solid-state reactions are either reversible or irreversible, and they may involve homogeneous single-crystal to single-crystal dimerisation or heterogeneous dimerisation, where the long-range order is destroyed.
We have investigated the photodimerisation reaction of α-styrylpyrylium trifluoromethanesulfonate (stypy(TFMS)). Upon excitation with visible light, the system changes colour, which can be reversed by heating above 350 K. This makes the system an ideal candidate for holographic optical storage devices based on organic materials. By combining high-resolution photocrystallographic experiments with picosecond time-resolved X-ray diffraction we have shown that the optical changes are directly related to the bond breaking/bond making mechanism shown in Figure 1.
![The [2+2] cycloaddition reaction of stypy(TFMS)](/files/live/sites/www/files/news/spotlight/spotlight100/spotlight100-fig1.jpg) |
|
Figure 1. The [2+2] cycloaddition reaction of stypy(TFMS). Conversion of the monomer (left) to the dimer (right) occurs upon excitation with visible light; the reverse reaction occurs above 350 K. |
High-resolution photocrystallographic studies of stypy(TFMS) single crystals were performed at the F1 and D3 beamlines of the Doris storage ring at Hasylab/DESY (Figure 2).
 |
|
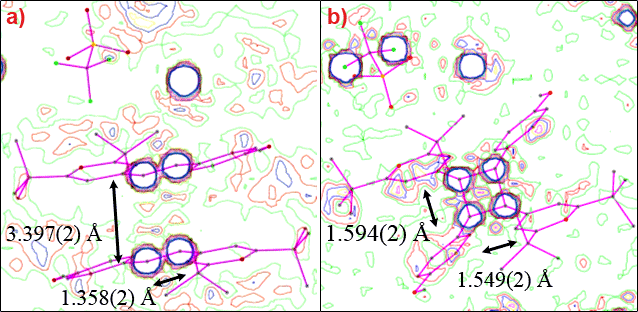
Figure 2. Electron density maps of a) the stypy(TFMS) red monomer phase and b) the yellow dimer phase. During photodimerisation, the distance between the two monomer molecules diminishes from 3.397(2) Å to 1.594(2) Å, while the 1.358(2) Å double bond becomes a 1.549(2) Å single bond. Besides flipping the phenyl residues toward the cyclobutyl ring, the cycloaddition reaction also causes a rotation of the TFMS anion. |
The picosecond time-resolved measurements were performed at ESRF beamline ID09B. In these ultrafast experiments, the photoreaction was initiated with 100 fs optical laser pulses that were synchronised with the time structure of the X-ray beam. A Ti:sapphire laser system with an OPG/OPA (optical parametric amplification) unit was used to generate 520, 550, and 575 nm excitation wavelengths. The chosen wavelengths guarantee a homogeneous reaction of the crystal. The X-ray sample geometry was set to grazing incidence and diffraction patterns were measured in rocking scan mode. The repetition frequency of the whole experiment was 986.3 Hz. Single X-ray pulses (100 ps long) from the storage ring 16 bunch mode were selected by a high-speed X-ray chopper.
These diffraction studies allowed the photoreaction in crystalline thin films to be monitored under experimental conditions where the transformation times were greatly enhanced. Figure 3 shows snapshots from a movie of the time-evolving (012) Bragg reflection of stypy(TFMS) crystals upon photoexcitation. Note, that the time-resolution of the experiment is limited by the synchrotron X-ray pulse width.
 |
|
Figure 3. Snapshots of the intensity variations of the (012) Bragg reflection upon pulsed laser excitation. |
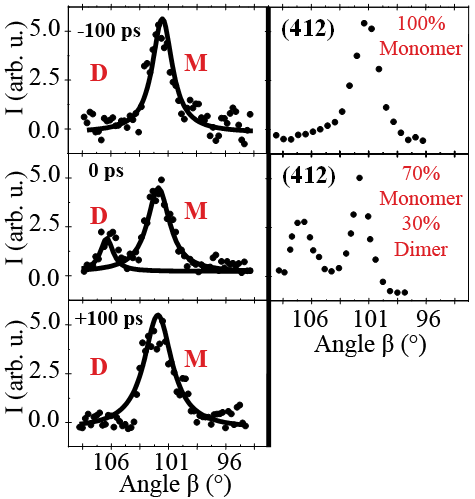
The data evaluation of the picosecond time-resolved crystallography studies reveals that stypy(TFMS) changes from a monomer state to a dimer product state. The formation of “short lived” crystalline domains with a mixture of these states has been realised in this study. Figure 4 compares the result of the picosecond time-resolved experiments (left) with “quasi-stationary” ones from reference samples (right) for the (412) Bragg reflection as an example. Around time zero, a well-defined dimer Bragg peak appeared, originating from the short-lived dimer phase. Under the experimental optical excitation conditions, the system undergoes a bulk transformation, deduced from the ultrafast formation and decay of Bragg peaks originating from the switching of whole domains to the dimer phase. Kinetic analysis based on the JMAK model (Johnson–Mehl–Avrami–Kolmogorov), reveals that the mechanism is not cooperative. The kinetics were deduced as a two-state process with changes of the molar fraction of the monomer to the dimer phase. The population of the states depends on the number of optical photons and the optical excitation wavelength. Depending on the temperature of these thin crystalline films, ultrafast time-resolved grazing incidence experiments proved the reversibility of the photoreaction with time constants below 30 ps. The present experiments indicate that the system reacts on time scales which are the fundamental limiting ones of two-quantum systems and therefore has the potential to be used as an ultrafast molecular switch.
 |
|
Figure 4. Left, ultrafast rise and decay of the (412) Bragg intensity of the dimer for laser/X-ray time delays of 100 ps, 0ps and 100 ps. The splitting of the (412) peak of the monomer and dimer phase permits monitoring the change of the alpha-angle of the monoclinic unit cell from 101.5° to 106°. Right, the (412) peak for the pure monomer and for a mixture of monomer and dimer in one crystal (70% monomer, 30% dimer). |
References
[1] M.D. Cohen, G.M.J. Schmidt, J. Sonntag, J. Chem. Soc., 2000 (1964).
[2] M. Irie, S. Kobatake, M. Horichi, Science 291, 176 (2001).
[3] S. Ohba, H. Hosomi, Y. Ito, J. Am. Chem. Soc. 123, 6349 (2001).
[4] V. Enkelmann, G. Wegner, J. Am. Chem. Soc. 115, 10390 (1993).
Principal publication and authors
J. Hallmann (a), W. Morgenroth (b,c), C. Paulmann (d), J. Davaasambuu (a), Q. Kong (e), M. Wulff (e), and S. Techert (a), Time-resolved X-ray diffraction of the photochromic α-styrylpyrylium trifluoromethanesulfonate crystal films reveals ultrafast structural switching, J. Am. Chem. Soc. 79, 131, 15018-15025 (2009).
(a) IFG Structural Dynamics of (Bio)chemical Systems, Max Planck Institute for Biophysical Chemistry, Goettingen (Germany)
(b) HASYLAB at DESY, Hamburg (Germany)
(c) Institut fuer Geowissenschaften, Facheinheit Mineralogie, Abt. Kristallographie, Universität Frankfurt (Germany)
(d) Mineralogisch-Petrographisches Institut, Universtität Hamburg (Germany)
(e) ESRF
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.