- Home
- Structural basis of lesion recognition by Rad14 (XPA)
Structural basis of lesion recognition by Rad14 (XPA)
Damage recognition mechanisms are a pivotal element in any DNA repair mechanism, since this step decides whether a DNA region should be repaired or not. To extend the knowledge of damage recognition strategies, the structures of Rad14 (XPA), an essential protein in nucleotide excision repair, have been analysed in the presence of different DNA damages.
Our DNA is constantly challenged by endogenous and exogenous sources. It is estimated that each cell in our body encounters approx. 10,000 lesions per day. Counteracting measures to maintain genomic integrity are thus essential. Among the different DNA repair systems, nucleotide excision DNA repair (NER) stands out due to its capability to recognise a vast variety of structurally diverse DNA damages. A defect in NER causes the disease xeroderma pigmentosum. One of the major hall-marks of this disease is the extreme sensitivity to sunlight associated with a highly increased risk of developing skin cancer. The entire repair process requires the presence of up to 30 different proteins, of which the XPA protein assumes a central role.
 |
|
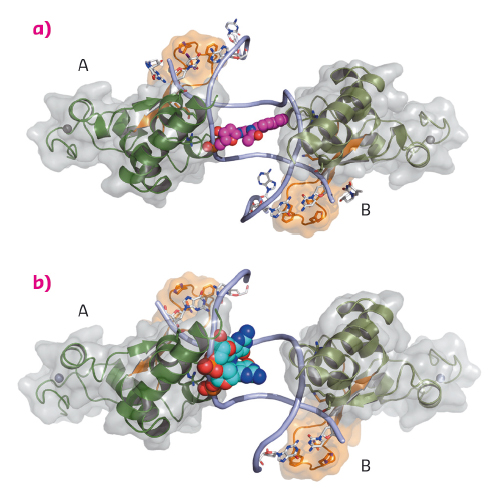
Fig. 127: The DNA binding domain of Rad14 bound to DNA. The different Rad14 monomers are labeled A and B. The finger motif is coloured in orange. For clarity only the DNA backbone is shown. The lesion is depicted in space-filling mode. (a) Rad14 bound to DNA containing a AAF-dG bulky adduct. (b) Rad14 bound to DNA containing a cisplatin 1,2-GG intrastrand crosslink. |
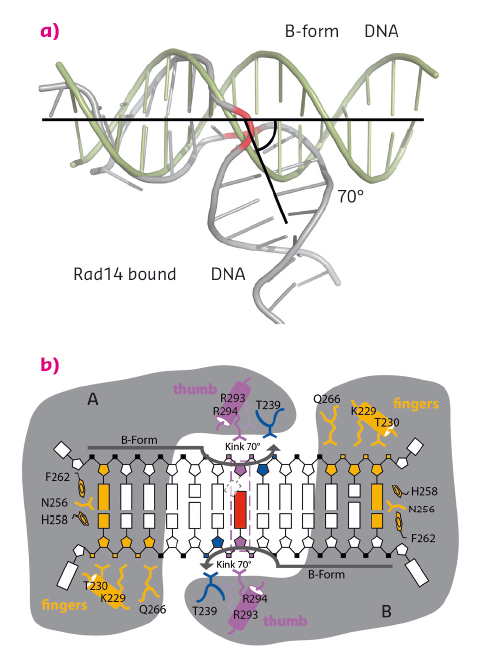
We solved crystal structures of the DNA binding domain of RAD14, the yeast homolog of XPA, in the presence of two different DNA damages: a cisplatin 1,2-GG intrastrand crosslink and a N-(deoxyguanosin-8-yl)-2-acetylaminofluorene (AAF-dG) bulky adduct. The crystallographic data were in part collected at the beamlines ID29 and ID23-2. The most striking feature of the two structures is the similar binding mode of Rad14 to the DNA (Figure 127) with two Rad14 monomers bound to the DNA forming a dimer on DNA without direct contact of the monomers. Moreover, the DNA in both structures showed a distinct kink of 70° at the site of the lesion (Figure 128a) which is stabilised by the two independent Rad14 monomers. Both molecules create a 13mer recognition motif with the lesion at the centre. The most prominent binding feature is a -hairpin in Rad14 that intercalates at the DNA ends which we termed the “fingers” motif. Whereas the fingers hold the DNA, stabilisation of the kink is achieved by a thumb consisting of Arg294 in-helix 7. The thumb binds to the DNA backbone at the site of the lesion thereby creating the central hinge where the DNA is kinked. Directly after the hinge, Thr239 further stabilises the kink via backbone interactions. The above described interactions are accomplished by each Rad14 monomer with the lesion being located in the centre between the two protein molecules (Figure 128b). The validity of this recognition complex in solution was confirmed using mass spectrometry. In addition, the DNA binding mode observed in the structure perfectly matches biochemical data obtained with XPA and different DNA substrates [1]. The DNA damage analysed in our structures are known to support either pre-kinked structures or allow DNA kinking that is not observed in regular B-form DNA. Thus, Rad14 seems to bind and test the DNA for bendability in order to verify the presence of a lesion in a lesion unspecific manner through a novel mechanism. Other damage binding proteins involved in NER like DDB2 or XPC/HR23B have also been shown not to engage a specific damage directly but rather sense backbone anomalies [2].
 |
|
Fig. 128: Rad14 kinks DNA. (a) Superposition of regular B-form DNA with Rad14 bound DNA. The bend angle is indicated. (b) Schematic overview of the Rad14 DNA binding mode with kinked DNA. |
Although XPA has not been directly implicated as a damage sensor, the structural and biochemical data obtained in our study may explain how NER might reach its high promiscuity towards different DNA lesions. The Rad14 crystal structures add another dimension to the mechanistic damage sensor portfolio revealing a mechanism for the specific recognition of kinked DNA structures in a sequence and lesion independent way.
Principal publication and authors
Structural insights into the recognition of cisplatin and AAF-dG lesion by Rad14 (XPA), S.C. Koch (a), J. Kuper (b), K.L. Gasteiger (a), N. Simon (a), R. Strasser (a), D. Eisen (a), S. Geiger (a), S. Schneider (c), C. Kisker (b) and T. Carell (a), PNAS 112, 8272–8277 (2015); doi: 10.1073/pnas.1508509112.
(a) Center for Integrated Protein Science at the Department of Chemistry, Ludwig-Maximilians Universität München, Munich (Germany) (b) Rudolf Virchow Center for Experimental Biomedicine, Institute for Structural Biology, University of Wuerzburg (Germany)
(c) Department of Chemistry, Technische Universität München, Garching (Germany)
References
[1] U. Camenisch, Nat Struct Mol Biol. 13, 278–284 (2006).
[2] J. Kuper, Curr. Opin. Struct. Biol. 22, 88–93 (2012).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.