- Home
- Molecular envelopes from protein powder diffraction data
Molecular envelopes from protein powder diffraction data
Deriving the crystal structure of protein molecules has been one of the key advances in understanding biology on the atomic length scale. The complexity of these molecules is so great that often several datasets must be used in combination to solve the crystallographic phase problem. In these cases, by carefully measuring the differences in diffracted intensities between different crystals, some containing additional heavy atoms, a sub-structure consisting of the positions of the added heavy atoms can often be found. Approximate values of the crystallographic phases can be computed from the heavy atom positions and observed intensities, leading to an electron density map.
Until recently, protein structures seemed to be too large to tackle using powder diffraction methods. Nevertheless, it is very appealing to be able to determine protein structures without having to produce large single crystals! Since the possibility of refining protein structures from powder data has already been shown [1], and that powder data can be used to solve simple molecular replacement problems [2], we wondered whether powder data would also be good enough for the de novo phasing of macromolecular structures.
Powdered samples of heavy atom derivatives for two model systems were prepared: uranium-derivatised porcine pancreatic elastase and chicken egg white lysozyme derivatised with a gadolinium complex (Gd-Hp-Do3A). High resolution powder diffraction data were collected at the beamlines ID31 and BM01. Comparison of these data with those collected from native samples shows small shifts in lattice parameters and changes in peak intensities indicative of the binding of the heavy atoms. Integrated peak intensities were extracted from the powder profiles and supplied for processing with software originally developed for single crystal data. In order to combat the peak overlap problem, multiple datasets were combined to exploit anisotropic lattice changes which lead to peak shifts between different diffraction patterns. For elastase these peak shifts were caused by radiation damage and for lysozyme they were induced by using samples prepared at different pH values.
In the case of elastase, the position of the uranium atom could be found from a difference Patterson map (Figure 77). The gadolinium positions in the lysozyme sample were found using the SHELXD software. In both cases the heavy atom positions found from powder data are fully consistent with earlier single crystal studies.
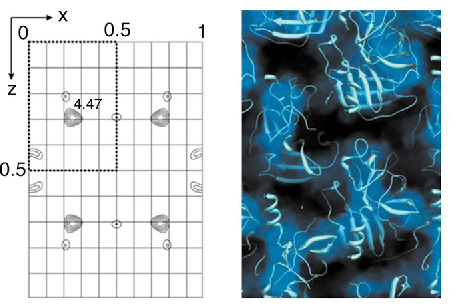 |
| Fig. 77: Difference Patterson map (left) and solvent mask (right) for the porcine pancreatic elastase [3]. |
Once the heavy atom positions had been determined, they were refined and phase calculations carried out using the SHARP software. Since there was only a single heavy atom derivative in each case, there is still an ambiguity in the determination of the phase. SHARP employs statistical methods and density modification techniques to resolve this.
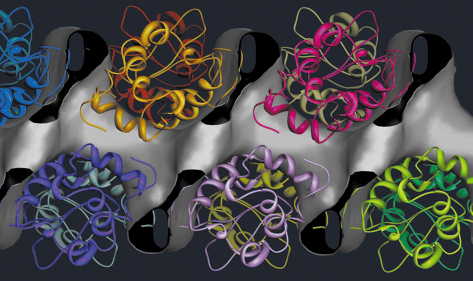 |
| Fig. 78: Solvent channel in lysozyme superimposed with the molecular structure. |
Electron density maps are produced at the end of the phasing procedure, as well as a solvent mask which delineates the regions of the unit cell occupied by protein and solvent respectively. Inspection of these maps indicates that the phasing procedure has been successful with a clear identification of the correct solvent boundaries within the unit cell (Figure 77 and Figure 78). Quantitatively, the derived phases show a good correlation with the true values up to a resolution of around 6 ÃÂ , indicating that using powder data in this way is still a relatively low resolution technique. Improvements in this area are anticipated with the development of phasing methods which take into account peak overlap in the powder data.
References
[1] R.B. Von Dreele, J. Appl. Cryst. 32, 1084-1089 (1999).
[2] I. Margiolaki, J.P. Wright, A.N. Fitch, G.C. Fox and R.B. Von Dreele, Acta Cryst., D61, 423-432 (2005).
[3] C. Besnard, F. Camus, M. Fleurant, A. Dahlström, J. P. Wright, I. Margiolaki, P. Pattison M. Schiltz. Z. Krist Submitted.
Principal Publication and Authors
J.P. Wright (a), C. Besnard (b), I. Margiolaki (a), S. Basso (a), F. Camus (b), A.N. Fitch (a), G. Fox (a), P. Pattison (a,b) and M. Schiltz (b), To be submitted.
(a) ESRF
(b) Laboratoire de Cristallographie, ÃÂcole Polytechnique Fédérale de Lausanne (EPFL) (Switzerland)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.