- Home
- News
- Spotlight on Science
- The effect on ligand...
The effect on ligand binding of displacing a single water molecule
07-12-2021
Crystallographic data from beamlines ID29 and ID30B shows that strategic placement of a single methyl group enables displacement of an unfavourable water molecule from the binding site of FKBPs. This results in the most potent FKBP-binders known to date, outperforming the natural products FK506 and rapamycin.
Share
An increasingly important class in medicinal research is the class of FK506-binding proteins (FKBPs). FKBPs have been associated with several diseases in recent years, including depression, obesity and chronic pain. In addition, microbial and parasitic pathogens contain FKBP-like proteins, called macrophage infectivity potentiators (Mips), which are important for the replication of these pathogens. Inhibition of FKBPs/Mips is therefore a treatment option for a variety of diseases. Since the natural ligands FK506 and rapamycin cause immunosuppression in complex with FKBPs, a need arose for ligands without immunosuppressive properties.
Understanding the underlying mechanisms that enable drugs to be highly potent is key in medicinal chemistry. One aspect is the binding of small molecules to their respective protein targets. Besides well-known interactions of the ligand with the protein, such as hydrogen bonds or Van-der-Waals interactions, the water network, surrounding both the ligand and the protein, plays a decisive role in determining binding properties. Replacement of energetically unfavourable (‘unhappy’) water molecules, while at the same time keeping energetically favourable (‘happy’) waters unperturbed, enables an improved water network and hence improved binding affinity.
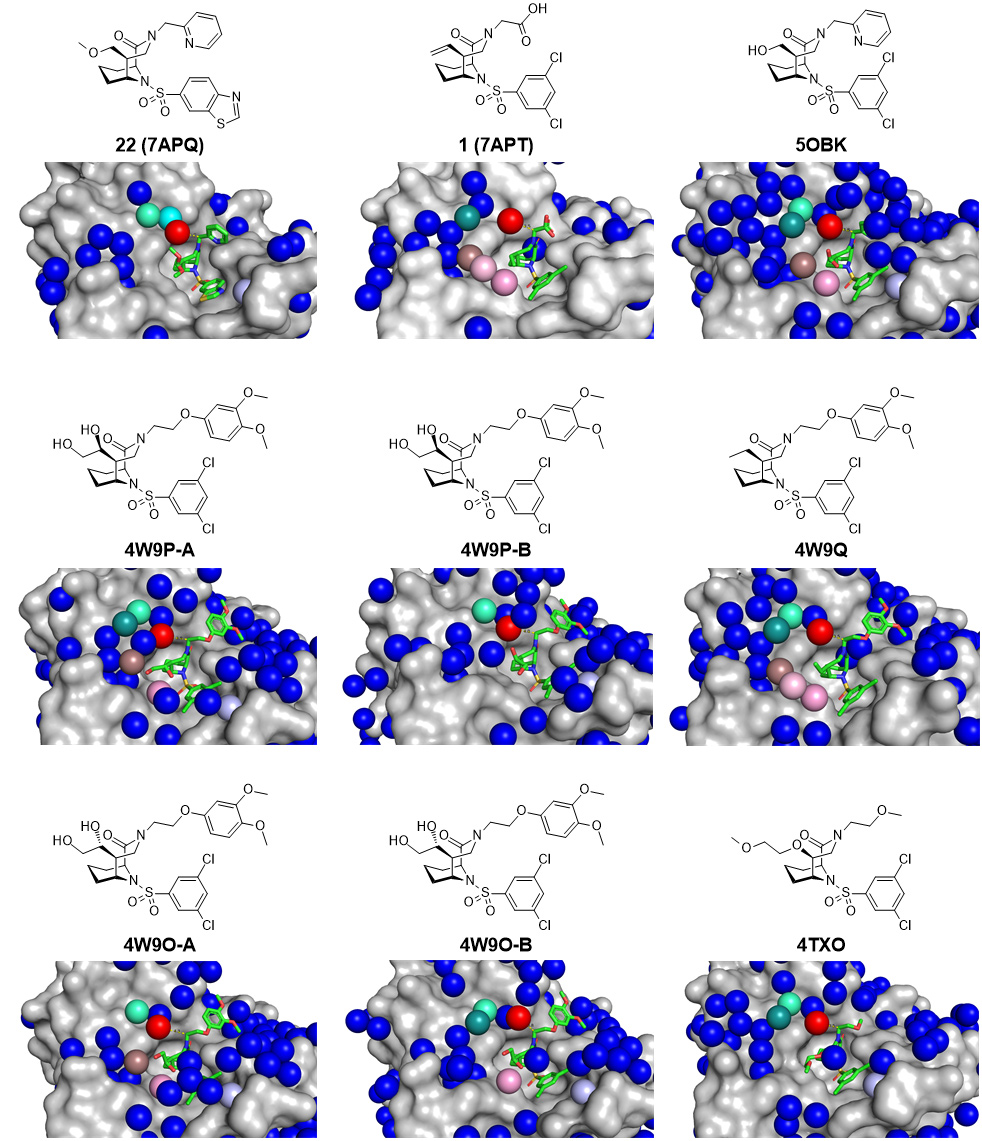
Fig. 1: Analysis of all available crystal structures (PDB-ID indicated) of FKBP51 (grey surface) containing bicyclic [4.3.1.] sulfonamide ligands (green sticks, chemical structures are given above the crystal structures). Crystallographically resolved water molecules are shown as blue spheres, the conserved water displaced by a methyl group in the α-position is highlighted in red, the distance to the α-position is indicated as yellow dotted lines in Å. Further conserved waters throughout the available crystal structures are highlighted in cyan, green, brown, pink and light pink.
Crystallographic data of FKBP51 in complex with several bicyclic [4.3.1.] sulfonamide ligands [1-5], were collected at beamline ID29. Evaluation of these crystal structures revealed several re-occurring water molecules (Figure 1). Analysis of the water sites with 3D-RISM disclosed a conserved ‘unhappy’ water molecule in all the FKBP-ligand crystal structures determined. The smallest chemical modification that might displace this ‘unhappy’ water is a methyl group. A small library, consisting of 28 bicyclic [4.3.1.] sulfonamides with a strategically placed methyl group, was thus synthesised and the compounds were tested for their binding affinity using fluorescence polarisation assays and isothermal calorimetry (ITC). All compounds with a methyl group in (S)-configuration displayed significantly improved binding affinity, while all (R)-isomers displayed decreased binding affinity. FKBP51 co-crystal structures determined at beamlines ID29 and ID30B confirmed that the (S)-methyl group displaced the targeted water molecule.
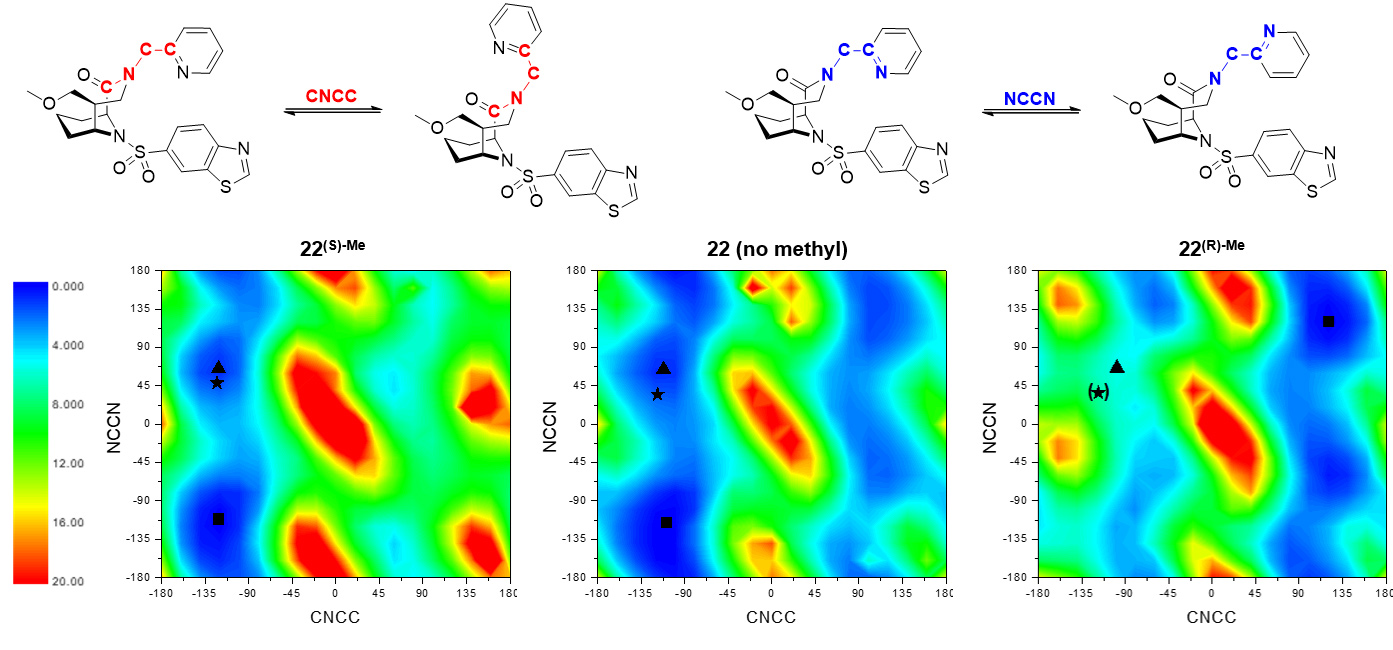
Fig. 2: DFT evaluation of conformations of the CNCC and NCCN dihedral angles next to the α-Me carbon for compounds 22, 22(S)-Me and 22(R)-Me [grid of 18x18]. Maps are coloured according to energy (blue: low energy; red: high energy; energies in kcal/mol). The black asterisk indicates the conformation found in the respective crystal structure (for 22(R)-Me: adopted from 22), a black triangle indicates the nearest local minimum, a black square indicates the global minimum.
Since methyl groups can profoundly affect binding energies by conformational effects, the influence of the methyl group on intrinsic conformational preferences was investigated. The rotational barriers for the NCCN and CNNC bonds were calculated by density functional theory, using the compounds 22, 22(S) Me and 22(R)-Me as a model system (Figure 2). The decreased binding of the (R)-isomer can be explained by an energetically unfavourable position of the methyl group, leading to an avoidance of the required conformation. The (S)-isomer shows a slightly higher occupancy of the conformation necessary for binding compared to the reference without methyl group; however, this does not fully explain the observed boost in affinity.
To further elucidate the enhanced affinity of the (S)-isomers, the thermodynamic signature was explored by ITC. This revealed that the affinity gain imparted by the additional methyl group was based exclusively on a gain in entropy, suggesting the displacement of the water molecule as major driver for the observed boost in affinity.
The most potent compounds of this study were tested for their cellular potencies and displayed significantly enhanced potency compared to their corresponding non-methylated analogues. Additionally, all compounds bound substantially better to intracellular FBKP12 compared to the prototypic FKBP ligand FK506. This is relevant because FKBP12 has been shown to repress BMP-signalling by binding to receptors of the ALK family and inhibition of FKBPs has been suggested as a potential treatment option for ALK-associated diseases.
Collectively, this research highlights the importance of the water network in ligand-protein complexes, and the developed compounds, as well as the associated findings, enable further advances in the development of therapies for FKBP-associated diseases.
Principal publication and authors
Picomolar FKBP inhibitors enabled by a single water-displacing methyl group in bicyclic [4.3.1] aza-amides, J.M. Kolos (a), S. Pomplun (b), S. Jung (c), B. Rieß (d), P.L. Purder (a), A.M. Voll (a), S. Merz (a), M. Gnatzy (a), T.M. Geiger (a), I. Quist-Løkken (e-g), J. Jatzlau (h), P. Knaus (h), T. Holien (e-g), A. Bracher (i), C. Meyners (a), P. Czodrowskic (c), V. Vera Krewald (a), F. Hausch (a), Chem. Sci. 12, 14758-14765 (2021); https://doi.org/10.1039/D1SC04638A
(a) Department of Chemistry, Technical University of Darmstadt (Germany)
(b) Leiden Academic Centre for Drug Research (LACDR), Leiden University (The Netherlands)
(c) Fakultät für Chemie und Chemische Biologie, Technische Universität Dortmund (Germany)
(d) Department of Chemistry, Technical University of Munich, Garching (Germany)
(e) Department of Clinical and Molecular Medicine, Norwegian University of Science and Technology, Trondheim (Norway)
(f) Department of Immunology and Transfusion Medicine, St. Olav's University Hospital, Trondheim (Norway)
(g) Department of Hematology, St. Olav's University Hospital, Trondheim (Norway)
(h) Institute for Chemistry and Biochemistry, Freie Universität Berlin (Germany)
(i) Research Department Cellular Biochemistry, Max Planck Institute of Biochemistry, Planegg (Germany)
References
[1] Y.S. Wang et al., J. Med. Chem. 56, 3922-3935 (2013).
[2] M. Bischoff et al., Org. Lett. 16, 5254-5257 (2014).
[3] M. Bischoff et al., Chemistry 26, 4677-4681 (2020).
[4] S. Pomplun et al., Angew. Chem. Int. Ed. 54, 345-348 (2015).
[5] S. Pomplun et al., J. Med. Chem. 61, 3660-3673 (2018).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.