- Home
- The crystal structure of human aquaporin 2
The crystal structure of human aquaporin 2
Maintenance of body water homeostasis is an essential part of human physiology. A key event in this is the kidney’s ability to regulate urine volume in response to dehydration. This takes place in the collecting duct through the action of aquaporin 2 (AQP2), a membrane-bound water channel, in the apical membrane of the principal cells. Upon dehydration, AQP2 is moved from intracellular storage vesicles to the apical membrane, thereby increasing its water permeability. This process, which is known as trafficking, involves the pituitary hormone vasopressin, which stimulates phosphorylation of the AQP2 C-terminus, thereby triggering its translocation. Defective trafficking of human AQP2 leads to nephrogenic diabetes insipidus (NDI), a water balance disorder characterised by large urine volumes (up to 20 L/day) and subsequent dehydration [1].
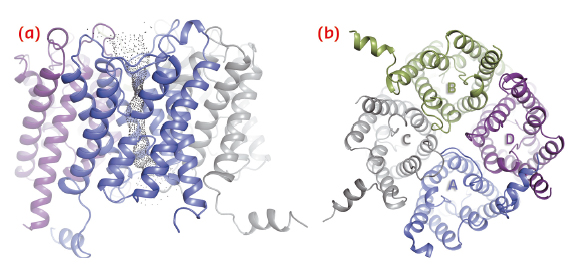
We have solved the crystal structure of human AQP2 at 2.75 Å resolution using data collected at ID14-4 (now ID30B). As seen in previous aquaporin structures, this reveals a tetrameric fold with each monomer comprising six transmembrane helices and two half-membrane spanning helices constituting a seventh pseudo-transmembrane segment (Figure 108). The water-translocating channel passes through the middle of each monomer in which water is transported in a single file while excluding ions and protons [2].
 |
|
Fig. 108: Structure of the AQP2 tetramer viewed in the plane of the membrane (a) and from the cytoplasm (b). |
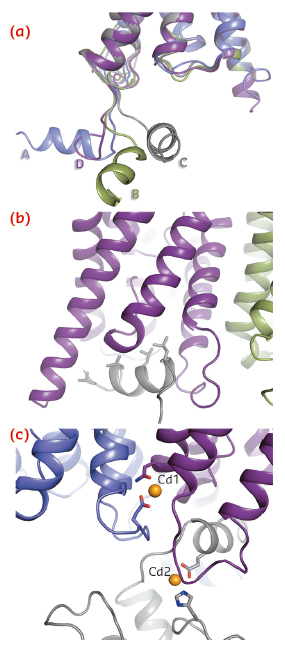
A striking difference between AQP2 and previous structures of mammalian aquaporins is the significant flexibility of the C-terminus. In particular, the C-terminal helix, which in previous aquaporin structures occupies a conserved position at the cytoplasmic interface, adopts four distinct new conformations (Figure 109a). In one monomer, the C-terminal helix interacts with the cytoplasmic interface of a symmetry-related AQP2 molecule (Figure 109b), thereby shielding four leucines aligned on the same side of the C-terminal helix from the cytoplasm. These leucine residues are responsible for the interaction between AQP2 and LIP5, a regulatory protein involved in targeting AQP2 for lysosomal degradation. The corresponding residues are also involved in the interaction between AQP0 and calmodulin [3]. We therefore suggest that exposed hydrophobic residues on the C-terminal helix are a common motif for protein-protein interactions amongst mammalian aquaporins.
 |
|
Fig. 109: Structural details of human AQP2. a) Overlay of four monomers showing the conformational variability of the C-terminus. b) Interaction between the C-terminal helix and the cytoplasmic interface of symmetry-related AQP2 molecules. c) Two cadmium ions can be seen per tetramer (Cd1 and Cd2) and are likely to represent calcium ion binding sites in vivo. |
An unexpected find in the AQP2 structure was the presence of two divalent cations per tetramer (Figure 109c), assigned as cadmium due to its presence during crystallisation, but likely to be replaced by calcium in vivo. The first binding site is located at the cytoplasmic interface between monomers A and D and causes a rearrangement of loop D, thereby exposing the binding pocket in which the C-terminal helix from the symmetry-related monomer binds (Figure 109b). The second calcium ion binds at the C-terminus and positions the C-terminal helix for this interaction. It thus seems that calcium ion binding may very well be important for protein-protein interactions involving the AQP2 C-terminus.
Due to its role in NDI, AQP2 is the most clinically studied member of the aquaporin family. Over fifty different AQP2 point mutations have been identified in patients suffering from NDI, most of which cause endoplasmic reticulum (ER)-retention. By obtaining the structure of AQP2, we now have a structural framework to help us understand why these mutations cause disease thereby opening the possibility of finding new pharmacological treatments. Furthermore, the crystal structure provides a platform for exploring membrane protein cellular sorting in general, including how this is guided by protein-protein interactions and how misfolded proteins are recognised by the ER quality control.
Principal publication and authors
A. Frick (a), U. Kosinska-Eriksson (a), F. de Mattia (b), F. Öberg (a), K. Hedfalk (a), R. Neutze (a), W. de Grip (c), P.M.T. Deen (b) and S. Törnroth-Horsefield (a, d), PNAS 111, 6305-6310 (2014).
(a) Department of Chemistry and Molecular Biology, University of Gothenburg (Sweden)
(b) Department of Physiology, Radboud University Medical Center, Nijmegen (The Netherlands)
(c) Department of Biochemistry, Radboud Institute for Molecular Life Sciences, Radboud University Medical Center, Nijmegen (The Netherlands)
(d) Department of Biochemistry and Structural Biology, Centre for Molecular Protein Science, Lund University (Sweden)
References
[1] P.M.T. Deen, M.A. Verdijk, N.V. Knoers, B. Wieringa, L.A. Monnens, C.H. van Os and B.A. van Oost, Science 264, 92-95 (1994).
[2] U. Kosinska Eriksson, G. Fischer, R. Friemann, G. Enkavi, E. Tajkorshid and R. Neutze, Science 340, 1346-1349 (2013).
[3] S.L. Reichow, D.M. Clemens, J.A. Freites, K.L. Nemeth-Calahan, M. Heyden, D.J. Tobias, J. E. Hall and T. Gonen, Nat. Struct. Biol. 20, 1085-1092 (2013).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.