- Home
- The structural basis of LPS recognition by the TLR4-MD-2 complex
The structural basis of LPS recognition by the TLR4-MD-2 complex
Lipopolysaccharide (LPS) is a major component of the outer membrane of Gram negative bacteria and is a potent activator of the human innate immune system. Minute amounts of LPS released into the blood stream by invading bacteria are an early sign of infection and prepare the immune system to counteract further infection. The immune response to LPS can also lead to fatal septic shock syndrome if the inflammatory response is amplified and uncontrolled. The core receptors recognising LPS are CD14, Toll-like receptor4 (TLR4), and MD-2. Binding of LPS to CD14 is enhanced by LBP (LPS-binding protein). Transfer of LPS from CD14 to TLR4-MD-2 heterodimers causes multimerisation of the receptor complex in the plasma membrane and triggers an intracellular signalling cascade. MD-2 is associated with the extracellular domain of TLR4 and is responsible for LPS binding. To date, ten members of the TLR family recognising a wide variety of microbial products have been identified in humans [1].
 |
|
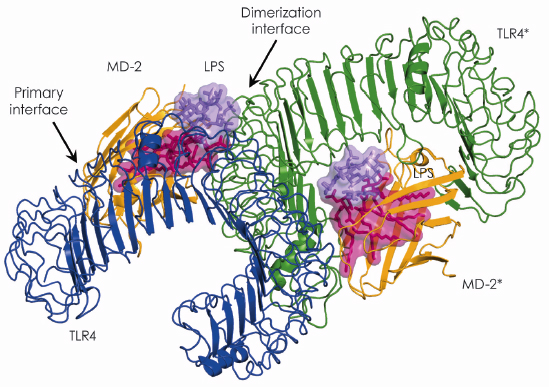
Fig. 115: Overall structure of the TLR4–MD-2–LPS complex. The primary interface between TLR4 and MD-2 is formed before binding LPS, while the dimerisation interface is induced by binding LPS. |
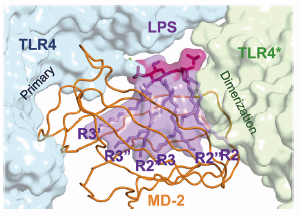
In 2007, using beamline ID14-2, we succeeded in determining the crystal structure of the TLR4-MD-2 complex [2]. The structure revealed that MD-2 interacts with the N-terminal and central area of the concave surface of TLR4. We also determined the structure of the TLR4-MD-2 in complex with Eritoran, an antagonist of TLR4-MD-2 and a candidate drug for severe sepsis. Eritoran binds to a large hydrophobic pocket in MD-2. At that time, however, we could not obtain diffraction-quality crystals of TLR4-MD-2 bound to LPS and therefore could only propose a model of the complex based on biochemical experiments. Early this year we finally succeeded in crystallising the TLR4-MD-2-LPS complex, and determined its structure at 3.1 Å resolution using the microfocus beamline ID23-2 [3]. Fortunately for us, the structure closely resembles the model we proposed two years ago. Binding of LPS induces the formation of a symmetrical dimer of two TLR4-MD-2-LPS complexes (Figure 115). The LPS bound to MD-2 directly bridges the two TLR4 molecules. The main dimerisation interface of MD-2 is located on the opposite side of the primary TLR4-MD-2 interface and interacts with the C-terminal domain of TLR4. Five of the six lipid chains in LPS are completely buried inside the MD-2 pocket but the sixth is partially exposed to the surface of MD-2 and forms a hydrophobic interaction with TLR4 (Figure 116). Hydrophilic side chains in the surrounding regions of MD-2 and TLR4 support the hydrophobic core of the interface by forming hydrogen bonds and ionic interactions. The two phosphate groups of lipid A reinforce this major dimeric interface by interacting with positively charged residues in TLR4 and MD-2.
 |
|
Fig. 116: The main dimerisation interface of the TLR4–MD-2–LPS complex. The inner core of LPS is omitted for clarity. |
Our crystal structure accounts for the structure-activity data for LPS accumulated over decades of research. Of the factors governing the immunological activity of LPS, the most important is the total number of lipid chains. Lipid A with six lipid chains has optimal inflammatory activity, while those with five lipid chains are ~100 fold less active, and those with four, such as Eritoran, completely lack agonistic activity. Based on our crystal structure we were able to predict that in LPSs with less than six lipid chains, all the chains move further into the pocket to fill the empty space, so that there is a substantial energetic penalty when they move back to the surface of MD-2 to dimerise with TLR4. Deletion of any of the two phosphate groups reduces endotoxic activity ~100 fold.
In summary, the crystal structure shows that multiple structural components of the TLR4-MD-2 receptor are involved in recognition of LPS. Further work is required to establish how these components interact with the structurally diverse LPS variants found in different bacterial species.
References
[1] N.J. Gay and M. Gangloff, Annu Rev Biochem 76, 141 (2007).
[2] H.M. Kim, B.S. Park, J.I. Kim, S.E. Kim, J. Lee, S.C. Oh, P. Enkhbayar, N. Matsushima, H. Lee, O.J. Yoo and J-O. Lee, Cell 130, 906 (2007).
[3] B.S. Park, D.H. Song, H.M. Kim, B.S. Choi, H. Lee and J-O. Lee, Nature 458, 1191 (2009).
Principal publication and authors
B.S. Park (a), D.H. Song (a), H.M. Kim (a), B.S. Choi (a), H. Lee (b) and J-O. Lee (a,c), Nature 458, 1191 (2009).
(a) Department of Chemistry, KAIST, Daejeon (Korea)
(b) Department of Biology, Chungnam National University, Daejeon (Korea)
(c) Institute of BioCentury, KAIST, Daejeon (Korea)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.