- Home
- Orientational ordering of non-planar phthalocyanines on Cu(111) revealed by X-ray standing wave experiments
Orientational ordering of non-planar phthalocyanines on Cu(111) revealed by X-ray standing wave experiments
The charge carrier transport across organic semiconductor-metal interfaces depends strongly on the energy level alignment of the two materials. To minimise the resulting energy barrier a controlled manipulation of the interface dipole (ID) would be very effective. In most cases, the complex interaction mechanisms of π-conjugated organic molecules and the rich phenomenology of adsorption geometries make this approach difficult. Detailed, multi-technique characterisations are often necessary to uncover the interplay between the structural and the electronic properties of the organic adlayer [1]. For instance, it has been shown that even planar molecules exhibit surprisingly different bonding distances and sometimes significant distortions of the entire molecule [2,3] because the covalent bonding between the metal substrate and the molecule can be altered by dispersion forces and electron transfer processes [4].
 |
|
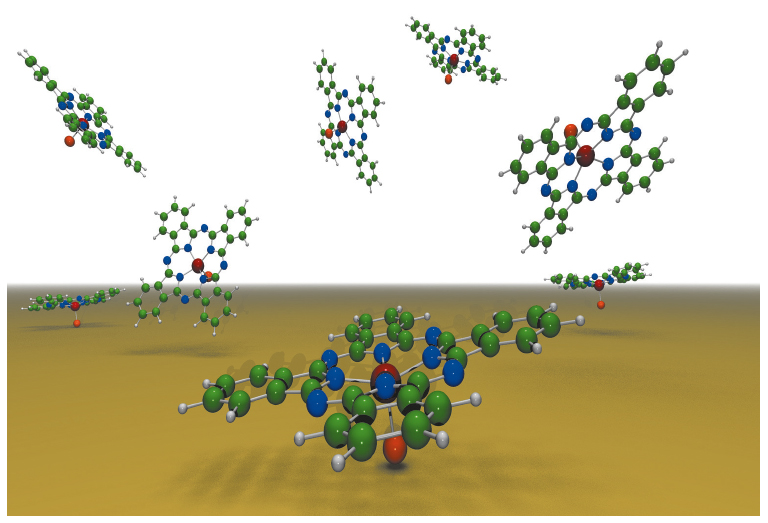
Fig. 39: Artist’s impression of GaClPc (Pc= C32N8H16) deposition on Cu(111). A preferential orientation of the molecules on the surface (either Cl-up or Cl-down) with an alignment of the intrinsic dipole moments is essential for the electronic structure of the entire system. X-ray standing wave experiments performed to measure the adsorption geometry and orientation of the molecules distinguish the different elements within molecules, i.e. Ga (red), Cl (orange), N (blue), and C (green). |
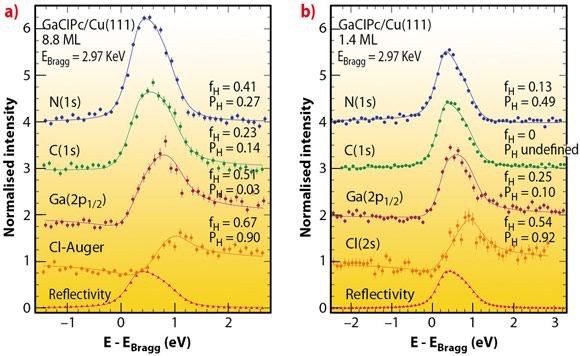
The adsorption behaviour of non-planar organic molecules with permanent dipole moments on metals is highly relevant to our aim of tuning the interface dipole. As these molecules can adsorb in different orientations (Figure 39), an (re-)alignment of the molecular dipoles on the surface would allow the work function and energy barrier at the interface to be influenced. In this context, we studied the orientational ordering of chlorogallium phthalocyanine molecules (GaClPc, Pc = C32N8H16) on Cu(111) using the X-ray standing wave (XSW) technique at beamline ID32. The element-specific photoelectron signals of the adsorbate were measured in a narrow energy range around EBragg=2.97 keV in order to determine the bonding distances dH. The results reveal that the majority of GaClPc molecules adsorb in a Cl-down configuration for coverages below one monolayer (Figures 40a and 41). Small deviations from the gas phase structure, as they were found in the XSW experiments, can be related to the interaction with the substrate and charge rearrangements at the organic-metal interface. For coverages above one monolayer (Figure 40b), the XSW data indicate a co-existence of the Cl-down and Cl-up configurations on the substrate. Eventually, the structural results for different coverage regimes – in particular the parallel/antiparallel orientation of the molecular dipoles – allow one to explain and analyse the observed work function change. DFT-based calculations confirm these findings and help to establish a quantitative picture of the GaClPc adsorption.
 |
|
Fig. 40: XSW data for GaClPc on Cu(111) taken for two different coverages in back-reflection geometry at an elevated temperature. The photon energy has been scanned around EBragg=2.97 keV to measure the photoelectron yield (circles) and reflectivity (triangles). Least-squares fits to the data (solid lines) give the coherent fraction fH and coherent position PH that are related to the adsorption geometry. |
 |
|
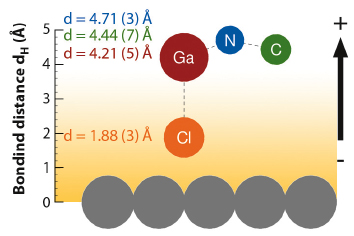
Fig. 41: Adsorption geometry of GaClPc as derived from the XSW data for 0.8 ML. The orientation of the molecular dipole leads to a decrease of the work function by ~0.3 eV, which is nearly half of the total adsorption induced change ∆Φ. Note that model calculations based on a statistical description of the GaClPc ensemble suggest that thermally activated librational modes of the molecules play an important role. |
This example illustrates the unique potential of XSW measurements, which give precise and chemically-resolved structural information even for relatively complex organic molecules, and thereby help to establish a better understanding of these systems.
Principal publication and authors
A. Gerlach (a), T. Hosokai (a), S. Duhm (b,c), S. Kera (c), O.T. Hofmann (d), E. Zojer (d), J. Zegenhagen (e) and F. Schreiber (a). Phys. Rev. Lett. 106, 156102 (2011).
(a) Universität Tübingen (Germany)
(b) Humboldt-Universität zu Berlin (Germany)
(c) Chiba University (Japan)
(d) Technische Universität Graz (Austria)
(e) ESRF (France)
References
[1] N. Koch, A. Gerlach, S. Duhm, H. Glowatzki, G. Heimel, A. Vollmer, Y. Sakamoto, T. Suzuki, J. Zegenhagen, J.P. Rabe and F. Schreiber. J. Am. Chem. Soc. 130, 7300 (2008).
[2] H. Yamane, A. Gerlach, S. Duhm, Y. Tanaka, T. Hosokai, Y.Y. Mi, J. Zegenhagen, N. Koch, K. Seki and F. Schreiber. Phys. Rev. Lett. 105, 046103 (2010).
[3] A. Gerlach, S. Sellner, F. Schreiber, N. Koch and J. Zegenhagen. Phys. Rev. B 75, 045401 (2007).
[4] L. Romaner, G. Heimel, J. L. Bredas, A. Gerlach, F. Schreiber, R.L. Johnson, J. Zegenhagen, S. Duhm, N. Koch and E. Zojer. Phys. Rev. Lett. 99, 256801 (2007).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.