- Home
- Plutonium redox chemistry on corroding steel surfaces
Plutonium redox chemistry on corroding steel surfaces
Nuclear waste has to be safely stored for geological time scales, even though current developments like fuel recycling and transmutation may substantially reduce the amount of waste in the future. The repository concepts of most countries utilising nuclear power foresee storage in the deep geological underground, which is commonly an anoxic environment unlike that exposed to our oxygen-rich atmosphere. Under such conditions, iron occurs predominately in its divalent oxidation state, and is able to reduce a number of radionuclides like selenium or uranium from higher to lower oxidation states, with important consequences for their chemical and migration behaviour.
Studying chemical reactions under such anoxic conditions requires the almost complete exclusion of oxygen from our oxygen-rich atmosphere down to levels of < 1 ppmv O2. While it is already a challenge to maintain such conditions from the laboratory to a public beamline for non- or low-radioactive samples, the effort required to do this safely with transuranium actinides is much higher. Plutonium in particular is of high interest since it occurs in relatively high quantity in burnt-up nuclear fuel and its nuclides have long half-lives (up to 80 million years) and a very high radiotoxicity. In this current study, we were able to study the redox chemistry of plutonium under anoxic conditions at the surface of three Fe(II)-bearing minerals for the first time. The mineral samples are representative of (anoxically) corroding steel containers and natural (anoxic) aquifers, they were the oxide magnetite, the sulfide mackinawite, and the hydroxo-carbonate chukanovite. The nanoparticulate mineral samples were prepared under anoxic conditions at the University of Grenoble, then reacted with Pu in an anoxic glovebox designed for radionuclides at KIT, transported from Karlsruhe to Grenoble in a special LN2 transport container developed by PSI, and finally measured at BM20, the HZDR-owned Rossendorf Beamline, where a custom-designed closed-cycle He cryostat as sample environment helped to exclude oxygen as well as to avoid photon-induced redox changes previously observed for plutonium. The purified 242Pu was provided by ITU.
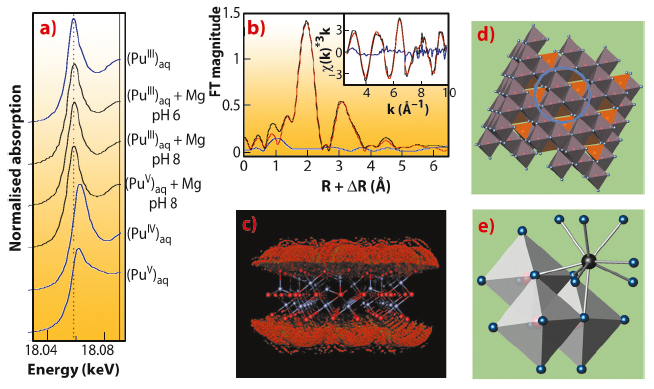
Using Pu-LIII edge XANES, we found that the initial Pu(V) aquocomplex with transdioxo coordination symmetry was reduced by magnetite at pH 6 and 8 to Pu(III) with tricapped trigonal-prismatic (TTP) coordination symmetry (Figure 91a). A relatively long equilibration period of 40 days as well as a cross-check with initial Pu(III) aquocomplex suggests that the trivalent oxidation state is thermodynamically stable. The radial structure function presented by the Fourier transform magnitude of the Pu-LIII EXAFS spectrum (Figure 91b) shows a strong symmetric peak typical for the quasi-equidistant arrangement of oxygen atoms in the TTP coordination shell of Pu(III). The smaller, asymmetric peak at about 3.25 Å (uncorrected for phase shift) is due to Pu-Fe backscattering as verified by Morlet wavelet analysis, while Pu-Pu backscattering could be excluded. To test the hypothesis of formation of an inner-sphere sorption complex at the magnetite surface, we applied a Monte-Carlo FEFF simulation method. Seven positions around a model slab of magnetite at the octahedrally terminated {111} face gave the lowest error and a very close fit of the experimental spectrum (Figure 91c,d). The matching structure is a trinuclear tridentate (with respect to surface Fe-octahedra), monomeric surface complex as shown in Figure 91e.
 |
|
Fig. 91: Pu-LIII XAS analysis of Pu redox processes at the magnetite/water interface. a) XANES. b) Best fit (red) of experimental EXAFS (black) as obtained by Monte-Carlo simulation. c) Probability of Pu positions at the surface of a magnetite slab (red: lower, green: higher likelihood). d) One of 7 best positions is marked with blue circle. e) Molecular structure of Pu(III) surface complex. |
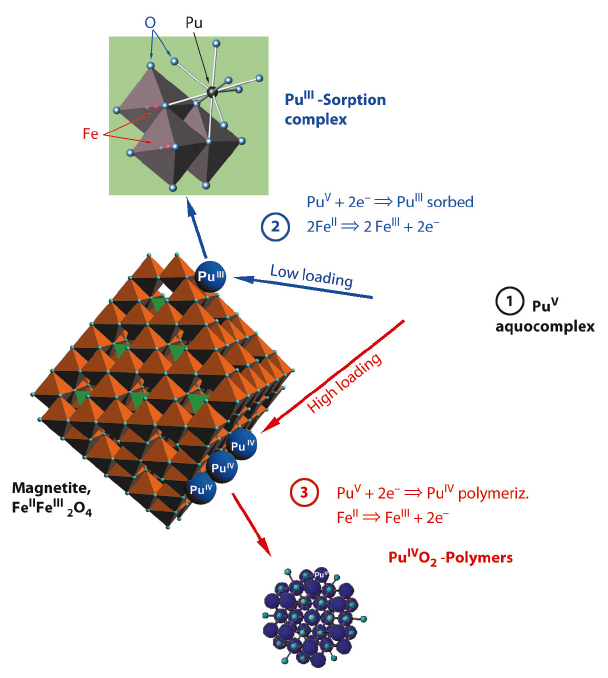
This is the first experimental evidence for the formation of such a trivalent Pu sorption complex. The few authors who previously studied Pu sorption reactions found formation of PuO2 colloids. By using higher surface loadings, we were able to replicate these older results (Figure 92, path 3). In terms of the safety assessment of nuclear waste repositories, both Pu(III) sorption and Pu(IV) precipitation have to be taken into account. Interestingly, both processes lead to similar, low Pu solubilities (<1010 M) – and both may be prone to colloidal transport under worst case scenarios.
 |
|
Fig. 92: Depending on the loading at the magnetite surface, aqueous Pu(V) (1) is either reduced to Pu(III), which forms a so-called inner-sphere sorption complex at the magnetite nanoparticles surface (2), or it is reduced to Pu(IV), which then polymerises to a PuO2-like solid (3). |
Principal publication and authors
R. Kirsch (a,b), D. Fellhauer (c), M. Altmaier (d), V. Neck (d), A. Rossberg (a), T. Fanghänel (c), L. Charlet (b) and A.C. Scheinost (a) Environ. Sci. Technol. 45, 7267–7274 (2011).
(a) Institut für Radiochemie, Helmholtz Zentrum Dresden Rossendorf, Dresden (Germany)
(b) Institute des Sciences de la Terre, Université Joseph Fourier, CNRS, Grenoble (France)
(c) European Commission, Joint Research Center, Institute for Transuranium Elements, Karlsruhe (Germany)
(d) Institut für Nukleare Entsorgung, Karlsruhe Institute of Technology, Karlsruhe (Germany)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.