- Home
- N-myristoyl transferase inhibitors as new leads to treat neglected diseases
N-myristoyl transferase inhibitors as new leads to treat neglected diseases
In the developing world, over 350 million people are at risk from neglected diseases. Existing drugs to treat diseases such as African sleeping sickness, Chagas’ disease, Leishmaniasis, and Malaria are often expensive, difficult to administer, unsafe and increasingly ineffective due to development of resistance. The parasites that give rise to these neglected diseases are well understood but this knowledge has not yet been translated into modern therapeutics for such diseases.
Protein N-myristoylation is a ubiquitous eukaryotic co- and post-translational modification and is required for membrane targeting and biological activity of many important proteins. The N-myristoylation reaction, i.e., the transfer of myristic acid from myristoyl-coenzyme A (CoA) to the amino group of N-terminal glycine residues within specific sequence contexts, is catalysed by the enzyme myristoyl-CoA: protein N-myristoyltransferase (NMT) [1]. NMT has been shown by gene knockout and RNAi to be essential for viability of the parasites Leishmania major and Trypanosoma brucei, therefore it has been identified as a viable target for the development of novel therapeutics [2]. A diversity-based library of 62000 compounds was therefore screened against NMT from T. brucei (TbNMT) yielding lead-like, chemically tractable hits. One hit series, based upon a pyrazole sulphonamide core, was developed to generate a lead compound DDD85646 that showed on-target potencies of 2 nM.
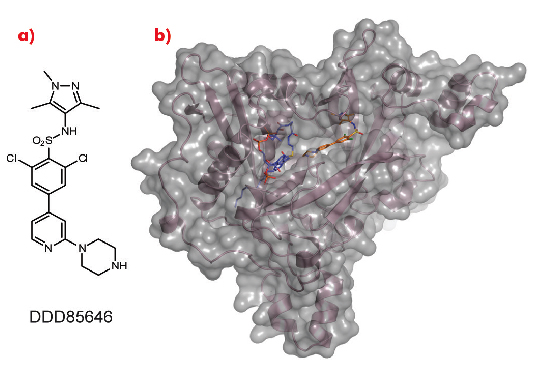
As a crystal structure of TbNMT has not yet been elucidated, we used Leishmania major NMT (LmNMT) as a structural surrogate due to its high level of sequence similarity to TbNMT, especially within the active site region. Our lead series of inhibitors also showed similar levels of potency against LmNMT as against TbNMT. The crystal structure of the ternary complex of LmNMT with bound co-factor myristoyl CoA and our lead molecule DDD85646 was then solved using data measured at beamline ID14-1. This structure, refined to a resolution of 1.6 Å, showed the pyrazole sulphonamide ligand DDD85646 to occupy the peptide binding cleft of LmNMT (Figure 98).
 |
|
Fig. 98: a) Chemical structure of DDD85646. b) the crystal structure of LmNMT (pink ribbon, grey surface) in complex with myristoyl coA (blue C atoms) and DDD85646 (gold C atoms). |
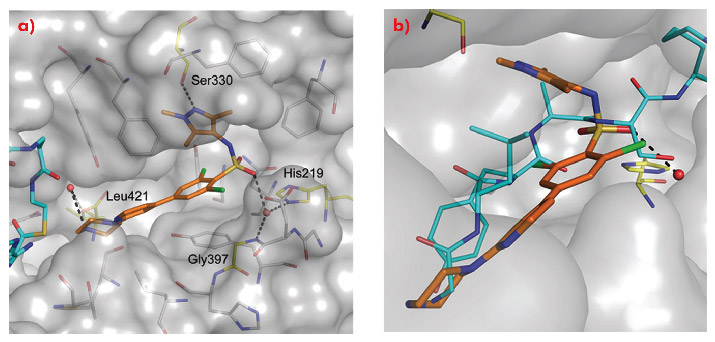
Comparison of the crystal structure of the ternary complex to that previously reported for Saccharyomyces cerevisiae NMT (ScNMT) in complex with a non-hydrolysable myristoyl CoA analogue and a prototypical substrate peptide GLYASKLA (PDB 1IID) shows that DDD85646 mimics the key recognition interactions between enzyme and peptide (Figure 99). The basic centre of the piperazine moiety mimics the N-terminal amide of the substrate peptide, interacting with the C-terminal carboxylate via a tightly bound water molecule. The sulphonamide forms a water bridged interaction with the backbone amide of Gly 397 and the side chain of His 219, with the water molecule occupying a similar position to the serine side chain of the substrate peptide. The sulphonamide moiety induces a pronounced kink in the molecule, allowing the trimethyl pyrazole moiety to pack into a hydrophobic cleft, forming a hydrogen bond with the side chain of Ser 330.
 |
|
Fig. 99: a) The mode of binding of DDD85646 (gold C atoms) to LmNMT (grey surface). Key water molecules are shown as red spheres, H-bonds as black dashed lines. Protein residues that interact with the ligand are highlighted (yellow C atoms) and labelled for clarity. b) DDD85646 mimics the binding mode of the substrate peptide GLYSAKLA (cyan C atoms). |
Regular access to ESRF beamlines has allowed us to use structural information to confirm that our novel small molecule DDD85646 binds in the active site of the target protein (NMT). Ongoing studies to identify novel templates and privileged binding pockets using fragment-based strategies are also facilitated by access to the high quality MX beamlines at the ESRF. This allows us to guide the development of chemical series to further optimise our target efficacy and selectivity to generate molecules suitable for clinical development.
Principal publication and authors
J.A. Frearson (a), S. Brand (a), S.P. McElroy (a), L.A.T. Cleghorn (a), O. Smid (a), L. Stojanovski (a), H.P. Price (d), M.L.S.Guther (a), L.S. Torrie (a), D.A. Robinson (a), I. Hallyburton (a), C.P. Mpamhanga (a), J.A. Brannigan (c), A.J. Wilkinson (c), M. Hodgkinson (d), R.Hui (e), W Qiu (e), O.G. Raimi (b), D.M.F van Aalten (b), R. Brenk (a), I.H. Gilbert (a), K.D. Read (a), A.H. Fairlamb (a), M.A.J. Ferguson (a), D.F. Smith (d) and P.G. Wyatt (a), Nature 464, 728-732 (2010).
(a) Division of Biological Chemistry and Drug Discovery, College of Life Science, University of Dundee (UK)
(b) Division of Molecular Microbiology, College of Life Science, University of Dundee (UK)
(c) Structural Biology Laboratory, Department of Chemistry, University of York (UK)
(d) Centre for Immunology and Infection, Department of Biology and Hull York Medical School, University of York (UK)
(e) Structural Genomics Consortium, University of Toronto (Canada)
References
[1] T.A. Farazi, G. Waksman and J.I. Gordon, J. Biol. Chem. 276, 39501-39504 (2001).
[2] H.P. Price, M.R. Menon, C. Panethymitaki, D. Goulding, P.G. McKean and D.F. Smith, J. Biol. Chem. 278, 7206-7214 (2003).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.