- Home
- Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex
Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex
Hsp90 (heat shock protein 90) is essential for cell viability in all eukaryotes and plays a central role in the maturation and activation of signalling proteins including steroid hormone receptors and protein kinases. The involvement of many Hsp90 client proteins in the development and progression of cancer has led to Hsp90 emerging as an exciting and key target for the development of new anti-tumour therapeutics. Activation of client proteins by the dimeric Hsp90 chaperone involves a range of co-chaperone proteins that complex with Hsp90 at different stages of the chaperone cycle and regulate and facilitate client protein activation [1]. One of the best-characterised client protein activation processes is that of steroid hormone receptors. During this process the steroid hormone receptor is bound to Hsp90 in a complex that is stabilised by the binding of the co-chaperone p23/Sba1 in an ATP dependent manner and allows the receptor to attain the activated conformation in which it can bind steroid hormone.
Despite initial uncertainty Hsp90 was identified as an ATPase. However the structural mechanism of this activity, and its role in Hsp90 chaperone function have been poorly understood and often contentious. From structural studies of individual domains of Hsp90, combined with biochemical and mutagenesis studies, we developed a mechanistic model in which binding and hydrolysis of ATP is coupled to a conformational cycle involving transient dimerisation of the N-terminal domains, which facilitates ATP turnover [2]. Biochemical evidence suggested that the p23/Sba1 co-chaperone functioned by binding selectively to this N-terminally dimerised conformation of Hsp90 and extended the lifetime of the closed ATP-bound state required for activation of a bound client protein.
 |
|
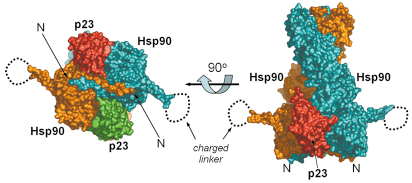
Fig. 61: Full-length hsp90-nucleotide-p23/sba1 shown as a molecular surface representation. The left handed twist of Hsp90 (blue and orange) can be seen (right), with the two p23/Sba1 (red and green) molecules symmetrically disposed on each side, contacting the N-terminal and middle domains. When the view is rotated 90° along the horizontal axis (left) the strand exchange between the two monomers is evident. The charged linker is disordered in the crystal. The crystal structure was refined to 3.1 Å resolution and data were collected at beamline ID29. The high intensity and stability of the beam was crucial for data acquisition. |
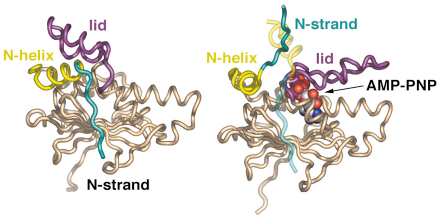
With the recent determination of the crystal structure of full-length Hsp90 in the closed state, in a ternary complex with a non-hydrolysable ATP analogue (AMP-PNP) and the p23/Sba1 co-chaperone, we have now been able to confirm this model and reveal the plethora of interconnected conformational changes that accompany ATP-binding (Figures 61 and 62). Central to the process is the movement of a segment in the N-terminal domain of Hsp90, the ‘lid’ region, which undergoes a very substantial restructuring on ATP-binding, folding over the mouth of the nucleotide binding pocket to interact with the ![]() -phosphate of the nucleotide. Lid closure exposes a hydrophobic patch on the N-terminal domains which forms a dimer interface. Accompanying this is a strand exchange between the two N-terminal domains which is also seen in other dimeric members of the GHKL ATPase superfamily including DNA gyrase B and MutL. The conformational changes in the N-domains also allow the docking of the middle segments of the chaperone and the formation of the active site at the interface of the N-domain and middle segments.
-phosphate of the nucleotide. Lid closure exposes a hydrophobic patch on the N-terminal domains which forms a dimer interface. Accompanying this is a strand exchange between the two N-terminal domains which is also seen in other dimeric members of the GHKL ATPase superfamily including DNA gyrase B and MutL. The conformational changes in the N-domains also allow the docking of the middle segments of the chaperone and the formation of the active site at the interface of the N-domain and middle segments.
 |
|
Fig. 62: Secondary structure diagram comparing the isolated N domain (left) and the AMP-PNP-bound state (right). Upon nucleotide binding the lid region closes over the active site, with considerable movement of the N-terminal helix and complete detachment of the N-terminal strand, which swaps into the other N domain of the dimer. |
The p23/Sba1 co-chaperone binds across the dimerised N-terminal domains of Hsp90, interacting with the middle segment of the chaperone and with the lid segment in its closed conformation. Intriguingly, the binding site of p23/Sba1 on the middle segment overlaps the binding site for another co-chaperone, Aha1 which greatly activates ATP turnover. Both co-chaperones cause restructuring of a catalytic loop in the middle segment of the chaperone allowing a key catalytic arginine residue to interact with the ![]() -phosphate of ATP and promote hydrolysis. However, because of its additional interaction with the closed conformation of the lid segment, p23/Sba1 slows turnover and release of the products, extending the lifetime of the closed state.
-phosphate of ATP and promote hydrolysis. However, because of its additional interaction with the closed conformation of the lid segment, p23/Sba1 slows turnover and release of the products, extending the lifetime of the closed state.
The structure of the full Hsp90-ATP-p23/Sba1 complex, along with previous crystal structures of Hsp90 domains and their complexes with other co-chaperone is providing an unparalleled level of insight into the detailed conformational biochemistry of this complex macromolecular system. The main challenge for the future lies in determining structures for Hsp90 complexes with client proteins so that the coupling of the Hsp90 ATPase cycle to client protein activation can finally be understood.
References
[1] L.H. Pearl and C. Prodromou, Annu Rev Biochem 75, 271-294 (2006).
[2] C. Prodromou et al., EMBO J. 19, 4383-4392 (2000).
Principal Publication and Authors
M.M. Ali, S.M. Roe, C.K. Vaughan, P. Meyer, B. Panaretou, P.W. Piper, C. Prodromou and L.H. Pearl, Nature 440, 1013-1017 (2006).
Section of Structural Biology, Institute of Cancer Research, London (UK)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.