- Home
- News
- Spotlight on Science
- X-rays reveal the...
X-rays reveal the energy storage mechanism of glow-in-the-dark materials
20-07-2020
The mysterious greenish light from glow-in-the-dark materials has fascinated many, including physicists and chemists puzzled by the working principle. A combination of optical and X-ray spectroscopy has now revealed the hiding place of the electrons driving the energy storage.
Share
Already in the early seventeenth century, a recipe circulated to turn certain volcanic minerals into glow-in-the-dark materials. Those compounds absorb and store part of the energy they receive when exposed to sunlight. This energy is then slowly released in the form of light, in a process that could last for hours. Unfortunately, the exact recipe got lost before science could offer an explanation. After an intermezzo in the early twentieth century, when self-luminous paints were powered by ionising radiation from radioactive sources, a new class of materials was found in the mid 1990s: strontium aluminate doped with small amounts of europium and dysprosium (SrAl2O4:Eu,Dy). This material can store energy when illuminated with blue light or near-ultraviolet radiation. It offers a long-lasting afterglow of tens of hours with a high initial brightness. This quickly led to safety applications like glow-in-the-dark signs indicating escape routes in case of an electrical power failure, used for example in planes and buildings. Other applications quickly followed, such as watch dials, small decoration stars, toys and even glowing cycling roads.
The underlying energy storage mechanism is complicated, which led to much debate in the growing scientific community working on those compounds. The first step in the process, the absorption of energy, is reasonably well established and in the benchmark compound, SrAl2O4:Eu,Dy, this task is performed by one of the lanthanides: europium. When in a divalent state (Eu2+), it absorbs blue or near-UV incident light, which lifts one of its electrons to an excited state. Normally, the electron returns almost immediately to the ground state and a greenish light is emitted. This is a similar process as the one used in most white light-emitting diodes (LEDs). In a persistent phosphor however, the electron is transferred to a crystallographic defect, or a trap, before this quick return can occur. These traps are introduced into the materials by incorporating another, trivalent lanthanide impurity, typically dysprosium (Dy3+), into the phosphor.
And this is where darkness entered, shrouding these phosphors in mystery. What is the role of the dysprosium co-dopant? Does it trap the electron, forming a rather exotic Dy2+ species, or does its size mismatch with the crystal lattice induce the necessary defects upon incorporation?
X-ray spectroscopy techniques are ideal tools to probe the valence state of the lanthanide ions inside such a persistent phosphor. Unfortunately, persistent phosphors are also strongly perturbed by the X-ray beam. When the beam is switched on, a bright luminescence emerges from the sample. We recorded the optical spectra of this X-ray-induced luminescence via an optical fibre, revealing characteristic emission bands from both europium and dysprosium. When the X-ray beam is switched off, the irradiated spot shows a strong afterglow, so the X-rays also lead to energy storage. The problem is that the traps are filled very fast, in the millisecond range for a high-brilliance beamline. A simple approach can however overcome this experimental constraint.
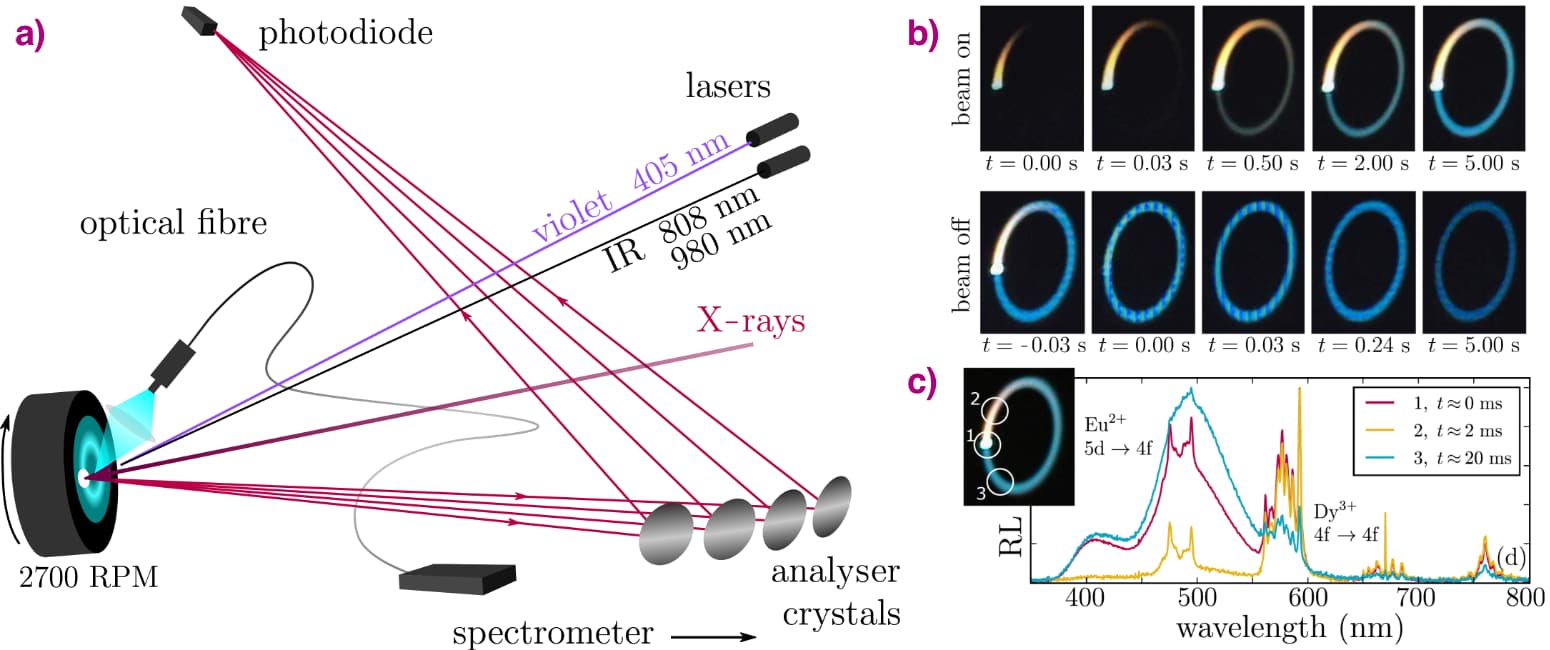
At beamline ID26, a pellet of the glow-in-the-dark phosphor was mounted on a spinning cooling fan, with the X-ray beam being positioned 5 mm off-axis which results in a strongly reduced average X-ray flux impinging on the sample surface. This approach does indeed prolong the time needed to fill the traps from a few milliseconds to 10 seconds, as shown by the slowly increasing afterglow intensity in Figure 1. To follow the movement of the electron, high energy resolution fluorescence-detected X-ray absorption spectroscopy (HERFD-XANES) was performed at the Eu L3 edge, monitoring the maximum of the Eu2+ and Eu3+ white lines on a second time scale. A reduction of the amount of Eu2+ was found, along with an equal increase of the amount of Eu3+. But more importantly, this change exhibits the same time dependency as the intensity of the light emission. This confirmed that the first step in the energy storage process indeed corresponds to the oxidation of the europium dopant.
 |
|
Figure 1. a) Schematic representation of the experimental setup: monochromatic X-rays strike the rotating sample, and the X-ray fluorescence is analysed. The radioluminescence (RL) is recorded by a fibre-coupled spectrometer and the sample can additionally be irradiated by violet or IR lasers. b) Camera images of a pristine rotating sample illustrate the charging and persistent luminescence of the bluish-green Eu2+ emission when the beam is switched on (top) and off (bottom). c) Emission spectra for different points along the racetrack, revealing spectral contributions from Dy3+ and Eu2+ in Sr4Al14O25:Eu,Dy. |
The exciting part was when the experiment was repeated at the Dy L3 edge. Initially, the sample contained only Dy3+ ions. When the X-ray beam was turned on, the white line indicative of Dy2+ appeared in the X-ray spectrum. Furthermore, the increase in Dy2+ concentration followed the same time profile as the light intensity and the increase of the Eu3+ (Figure 2). This demonstrated that the dysprosium co-dopants indeed act as the trapping centre of the electron originating from the europium luminescent centre, solving the longstanding question concerning the chemical nature of the traps. The glow-in-the-dark emission follows quickly after the thermally-assisted release of the trapped electron, oxidising the Dy2+ back to Dy3+.
 |
|
Figure 2. High energy resolution fluorescence-detected X-ray absorption near edge structure (HERFD-XANES) spectra for the (a) Eu and (b) Dy LIII edges upon violet (coloured line) or IR (black line) irradiation (top), compared to conventional XANES of reference spectra (middle) and the associated difference spectra (coloured lines) with their fit (black line) (bottom). (c) Time evolution of the lanthanide oxidation states, relative to the total [Eu]+[Dy] content from HERFD-XANES. Simultaneously, the luminescence is probed for d) the Eu2+ and e) the Dy3+ emission. The X-ray beam is switched on at time t = 0 while the sample spins at 2700 rpm. The key point is that the evolution of the Eu2+ light output follows the change in oxidation state of the lanthanide ions. |
The experimental approach – which can be extended by adding lasers to further control the number of trapped electrons – can now be used to investigate the role played by other defects and their interaction with the dysprosium ions. The good news is that only a few percent of the dysprosium ions participate in the trapping process, so there is room for improvement in the energy storage capacity. Much brighter glow-in-the-dark compounds could enable more demanding applications, following a better understanding of the delicate interactions in these persistent phosphors. Future experiments with these materials will make use of the much brighter X-ray beams of the ESRF-EBS now that a method has been developed to harness their power.
Principal publication and authors
Identification of Dy3+/Dy2+ as electron trap in persistent phosphors, J.J. Joos (a,b), K. Korthout (a,b), L. Amidani (c,d), P. Glatzel (c), D. Poelman (a,b), and P.F. Smet (a,b), Physical Review Letters (2020); DOI: 10.1103/PhysRevLett.125.033001.
(a) LumiLab, Department of Solid State Sciences, Ghent University (Belgium)
(b) Center for Nano- and Biophotonics (NB Photonics), Ghent University (Belgium)
(c) ESRF
(d) Current address: Helmholtz-Zentrum Dresden-Rossendorf, Dresden (Germany)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.