- Home
- Trapping a sliding clamp in action: how MutS activates MutL in DNA mismatch repair
Trapping a sliding clamp in action: how MutS activates MutL in DNA mismatch repair
DNA mismatch repair protects the genome from replication errors. In this process, the MutS protein activates MutL upon recognition of a mismatch and ATP binding. The crystal structure of the transient complex between MutS and the LN40 N-terminal domain of MutL was determined using a site-specifically cross-linked complex. This allowed analysis of the large conformational changes associated with sliding clamp formation. It showed why both mismatch and ATP are required for complex formation and how this step results in loading of MutL onto DNA. Validation by FRET, mutagenesis and stopped-flow kinetic analysis revealed the necessity of this step for DNA mismatch repair.
DNA mismatch repair (MMR) plays a crucial role in maintaining genome stability. Defects in the mismatch repair proteins in humans predispose them to Lynch syndrome (or hereditary non-polyposis colorectal cancer) and are associated with a variety of sporadic cancers. Upon replication, DNA mismatch repair is initiated by recognition of a mismatch or an unpaired base by MutS (in Escherichia coli) or its MSH homologues (in humans). Initial recognition of the mismatch is followed by an ATP-dependent conformational change of MutS into a sliding clamp state that can be recognised specifically by the next protein in the mismatch repair cascade, MutL (or its homologues). Intriguingly, both MutS and MutL are critical for correct repair and germline mutations in the human homologues of either of these proteins lead to cancer predisposition. How the transient sliding clamp of MutS promotes MutL homologue recruitment and activation of repair has long been unclear.
Here we have studied the sliding clamp state and the contribution of MutL to the mismatch repair initiation. The difficulty for structural analysis was the highly transient nature of the complex, requiring both mismatch and ATP, but causing a movement away from the mismatch. To trap this transient state we have made use of cysteine-free variants produced in the laboratory of P. Friedhoff in Giessen.
A highly specific cross-linked complex of MutS and MutL could be generated between specific single cysteines that would allow cross-linking of MutS to MutL only in the presence of a mismatch and ATP [1]. By scaling up the production of this cross-linked product and removal of flexible regions, we were able to obtain diffraction quality crystals of MutS (1-800) cross-linked to the LN40 domain of MutL. Data were collected at beamline ID29 using a Pilatus 6M detector, resulting in a 4.7 Å dataset that allowed structure solution.
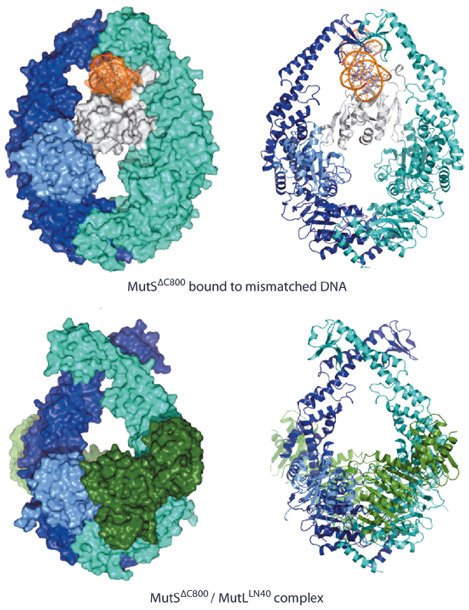
In the structure of the complex, we observe that MutS has made large scissor-like movements relative to the mismatch bound state [2] (Figure 135), tilting about the ATPase domains. This pushes DNA down into a new channel, such that MutS can slide along the DNA. Meanwhile, the N-terminal connector domain has rotated to contribute to a compound MutL interface, together with the ATPase domain of the other MutS subunit. We were able to validate the observed conformation as the sliding clamp by FRET analysis of fluorophores linked at different sites using the single cysteine variants of MutS and MutL. The observed interfaces could be validated by mutagenesis using MMR complementation and SPR binding studies. The data showed that both MutS subunits were required for stable interaction with MutL explaining why MutL requires the sliding clamp for binding.
 |
|
Fig. 135: Comparison of MutS DNA recognition and sliding clamp state. Top: Mismatch recognition state of MutS, shown in cyan and dark blue, N-terminal domain of subunit A, shown in light blue (connector domain) and white (mismatch recognition domain), DNA, shown in orange. Bottom: Crystal structure of MutS (colouring as above) complexed with the LN40 domain of MutL (dark green). Van der Waals surface and cartoon representations, left and right respectively. |
The new structure explains how the MutS sliding clamp promotes loading of MutL onto DNA, to activate downstream effectors. Using stopped-flow analysis, we could show that this was a slow step, after the fast recognition of the DNA. Using in vitro nuclease assays as well as in vivo complementation assays, we could show that this loading is essential for DNA mismatch repair. We thus elucidate a crucial mechanism that ensures that MMR is initiated only after detection of a DNA mismatch.
Principal publication and authors
MutS/MutL crystal structure reveals that the MutS sliding clamp loads MutL onto DNA, F.S. Groothuizen (a), I. Winkler (b), M. Cristóvão (b), A. Fish (a), H.H.K. Winterwerp (a), A. Reumer (a), A.D. Marx (b), N. Hermans (c), R.A. Nicholls (d), G.N. Murshudov (d), J.H.G. Lebbink (c), P. Friedhoff (b) and T.K. Sixma (a), eLIFE 4, e06744 (2015); doi: 10.7554/eLife.06744.
(a) Division of Biochemistry, Netherlands Cancer Institute, Amsterdam (The Netherlands)
(b) Institute for Biochemistry, Justus-Liebig-University, Giessen (Germany)
(c) Department of Genetics, Erasmus Medical Center, Rotterdam (The Netherlands)
(d) Structural Studies, MRC-LMB, Cambridge (UK)
References
[1] I Winkler et al., J. Biol Chem. 286, 17326-37 (2011).
[2] M.H. Lamers et al., Nature 407, 711-717 (2000).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.