- Home
- Converging peptide- and DNA-nanotechnology: molecular self-assembly of peptide nucleic acids
Converging peptide- and DNA-nanotechnology: molecular self-assembly of peptide nucleic acids
A new family of nanostructures sharing properties of both peptide and DNA was discovered. Peptide nucleic acids containing a peptide-like backbone and nucleic acid side-chains has been found to efficiently form ordered assemblies of unique optical properties. X-ray crystallography demonstrated the combination of stacking interactions together with canonical Watson-Crick base-paring as observed in double-helix DNA.
Biology serves as a major inspiration for the assembly of ordered structures at the nanoscale. Two main directions for the development of bio-inspired nanomaterials are based either on the use of protein and peptide building blocks or, alternatively, on the use of DNA. Polypeptide structures have the advantage of structural integrity and robustness while nucleic acids have the advantage of specific molecular recognition between complementary bases.
Our group has been extensively involved in the study of molecular self-assembly by extremely short peptide fragments. We demonstrated in 2003 that simple dipeptides contain all the molecular information needed to form ordered nanostructures [1]. Furthermore, peptide assemblies have been shown to exhibit remarkable physical properties including high mechanical rigidity, luminescence, piezoelectricity, and semiconductivity [2]. The dipeptide assemblies act as supramolecular polymers including a clear phase transition governed by Ostwald’s rule of stages [3] and thus may be compared to the polyamide covalent polymers mentioned above.
Parallel to the work on peptides, the field of DNA nanotechnology, pioneered by N.C. Seeman in the 1980s [4], is rapidly advancing and complex architectures at the nanoscale can be obtained.
We have sought to converge the two distinct fields of DNA and peptide self-assembly by using an artificially synthesised polymer that has attributes of both chemical families. This material, termed PNA (peptide nucleic acid) has the backbone of a peptide, but the residues of the DNA. This family of molecules that were developed in the 1990s are mainly used for gene regulation [5].
As we knew that dipeptides represent a minimalistic assembly unit, we synthesised 16 different di-PNAs with all possible combinations of the adenine (A), cytosine (C), guanine (G), and thymine (T) bases. We demonstrated the ability of very short di-PNA building blocks to self-assemble into ordered architectures. Intriguingly, only guanine containing building blocks were able to form ordered self-assembled structures. This is very interesting as guanine is readily used by nature to form physical colours and reflectors by molecular self-assembly.
 |
|
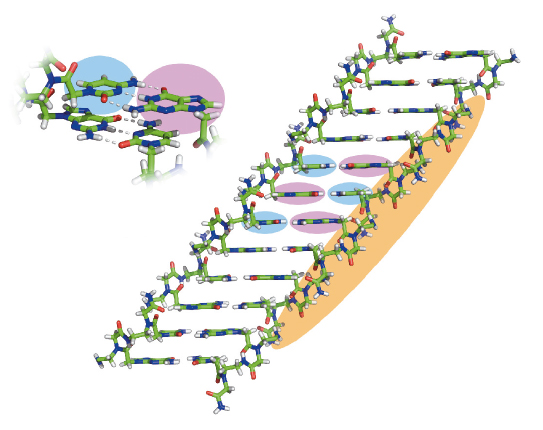
Fig. 118: The crystal structure of PNA assemblies. The atomic structure of the GC di-PNA shows that the bases are engaged in canonical Watson-Crick hydrogen bonding and that the aromatic bases stack in continuous chains where the G (pink background) and C (blue background) bases alternate along the length. |
The most interesting assemblies were of GC di-PNA that contain complementary cytosine and guanine that could undergo hydrogen bonded base-pairing (Figure 118). We found that in vitro the building blocks could self-associate to grow structures with an elongation rate 20 times faster than that of a microtubule. The optical properties of the newly characterised assemblies were also unique. We observed a clear fluorescent property of the assemblies with a very extensive red-edge excitation shift (REES) at the visible range of the electromagnetic spectrum (Figure 119). This phenomenon is often associated with dense structures such as glassy materials or graphene oxide and is exceptional for organic self-assembled structures.
 |
|
Fig. 119: The PNA assemblies exhibit a red edge excitation shift with a broad range of emission wavelengths at the visible region. One bright-field and five fluorescence images of the same microscopic field of GC di-PNA structures are shown. Each fluorescence image was taken with different excitation and emission filters: (excitation/emission) 387 nm/440 nm; 485 nm/525 nm; 537 nm/578 nm; 560 nm/607 nm; 650 nm/684 nm (from left to right). |
We used the energy tuning capabilities of beamline ID29 to collect X-ray data on thin, needle-like crystals of the GC di-PNA. By bringing the energy down to 15.59 eV, atomic resolution data could be measured. The resulting crystal structure shows that the GC bases form classical Watson-Crick hydrogen bonding and that the aromatic bases p stack in long, contiguous chains where the G and C bases alternate along the length of the stack.
The very unique architecture that results from both the peptide-like and DNA base pairing results also in unpredicted physical properties. The technological utilisation of this new family of self-assembled materials is now being explored.
Principal publication and authors
Light-emitting self-assembled peptide nucleic acids exhibit both stacking interactions and Watson–Crick base pairing, O. Berger (a), L. Adler-Abramovich (a), M. Levy-Sakin (a), A. Grunwald (a), Y. Liebes-Peer (a), M. Bachar (a), L. Buzhansky (a), E. Mossou (b,c), V.T. Forsyth (b,c), T. Schwartz (a), Y. Ebenstein (a), F. Frolow (a), L.J.W. Shimon (d), F. Patolsky (a) and E. Gazit (a), Nature Nanotechnology 10, 353–360 (2015); doi: 10.1038/nnano.2015.27.
(a) Tel Aviv University (Israel)
(b) Partnership for Structural Biology, ILL, Grenoble (France)
(c) Faculty of Natural Sciences, Keele University (UK)
(d) Weizmann Institute of Science, Rehovot (Israel)
References
[1] M. Reches and E. Gazit, Science 300, 625–627 (2003).
[2] L. Adler-Abramovich and E. Gazit, Chem. Soc. Rev. 43, 6881–6893 (2014).
[3] A. Levin et al., Nature Commun. 5, 5219 (2014).
[4] N.C. Seeman, J. Theor. Biol. 99, 237–247 (1982).
[5] P.E. Nielsen et al., Science 254, 1497–1500 (1991).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.