- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2013
- Structural biology
- Crystal structure of the entire respiratory complex I
Crystal structure of the entire respiratory complex I
Mitochondria are “cellular power plants”, containing a chain of respiratory complexes that utilise NADH (mainly) and oxygen to pump protons across the membrane. This proton-motive force is used to produce the universal energy currency ATP, by ATP synthase, a turbine-like molecular machine. Another example of a molecular machine is complex I, the first and largest enzyme in the respiratory chain. It transfers two electrons from NADH to quinone, coupled to the translocation of four protons across the membrane. Mitochondrial complex I consists of 44 subunits, whilst the prokaryotic enzyme is simpler, usually consisting of 14 “core” subunits with a total mass of about 550 kDa. It represents an important ‘minimal’ model of human complex I.
Defects, mainly related to complex I, in mitochondrial DNA are one of the most common types of human genetic disorder. So far there is no treatment for these debilitating diseases, which include neurological impairment, deafness, blindness, muscle weakness and cardiovascular disease. To understand the molecular basis of these conditions and as a starting point for drug development, we need to know the structure of the proteins involved. Structures are also necessary, of course, in order to understand the fundamental mechanistic principles of molecular machines.
Complex I, due to its sheer size, resisted structure determination efforts for a long time. Our earlier crystal structures of the hydrophilic domain of complex I from Thermus thermophilus established the electron transfer pathway from NADH through the flavin mononucleotide (FMN) and seven iron-sulfur (Fe-S) clusters to the putative quinone binding site [1]. Recently we have determined the atomic structure of the membrane domain of E. coli complex I (220 kDa, 6 subunits, 55 TM helices) at 3.0 Å resolution [2]. The structure revealed many unexpected features of the protein fold, such as the face-to-back arrangement of symmetry-related domains in the three similar antiporter-like subunits, the formation of a single proton-translocation channel from two half-closed channels found in each of these domains, and the central role of lysine residues (rather than usual carboxylates) in the proton channels.
 |
|
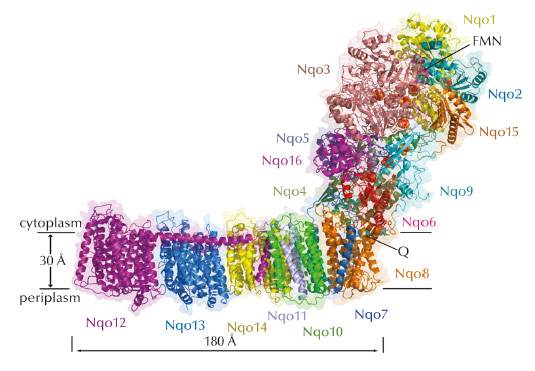
Fig. 46: Structure of the entire complex I from T. thermophilus. FMN and Fe-S clusters are shown as magenta and red-orange spheres. |
We have now determined the structure of the entire complex I from T. thermophilus (536 kDa, 16 subunits, 9 Fe-S clusters, 64 TM helices) to 3.3 Å (Figure 46). This is the largest asymmetric membrane protein structure so far determined, and solving it was a major technical challenge. Crystals of the entire complex are twinned, and so, to overcome a problem of model bias, we also crystallised and solved the structure of the isolated membrane domain of T. thermophilus complex I. The core fold of subunit Nqo8 (previously missing from structures) is, unexpectedly, similar to a half-channel of the antiporter-like subunits. Small subunits nearby form a linked second half-channel, thus completing the fourth proton translocation pathway (dubbed E-channel due an abundance of glutamates), in addition to the channels in three antiporter-like subunits. The quinone-binding site is unusually long, narrow and enclosed. The quinone head group binds at the enclosed end of this chamber, near cluster N2. This means that quinone, uniquely, moves about 15 Å out of the membrane in order to accept electrons. Such an unusual binding site allows the protein to control quinone protonation and to use the redox energy of charged species to drive conformational changes. The quinone chamber is linked to the fourth proton translocation channel by a “funnel” of charged residues. This link continues over the entire membrane domain as a remarkable flexible central axis of charged and polar residues.
 |
|
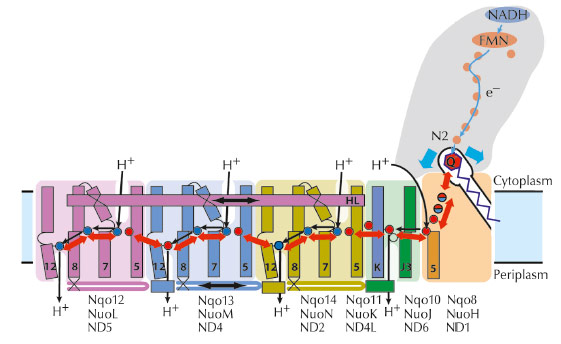
Fig. 47: Proposed coupling mechanism of complex I. Upon reduction, negatively charged quinone initiates conformational changes in the E-channel (Nqo8/10/11). These propagate to the antiporter-like subunits via the central axis (red arrows) of charged and polar residues located around flexible breaks in key transmembrane helices (numbered). Helix HL and the bH element (labelled) help coordinate conformational changes. Key charged residues are indicated by red circles for Glu and blue circles for Lys/His. Conformational changes drive appropriate changes in the pKa’s and solvent exposure of key residues so that net result is the translocation of four protons per cycle. |
The structure suggests the likely mechanism of coupling between the electron transfer in the hydrophilic domain and proton translocation in the membrane domain through long-range conformational changes (Figure 47). It somewhat resembles a steam engine with its coupling rods linking parts of the machine and is Nature’s compliment to the turbine-like ATPase.
Principal publication and authors
R. Baradaran (a), J.M. Berrisford (a,b), G.S. Minhas (a) and L.A. Sazanov (a), Nature 494, 443-8 (2013).
(a) Medical Research Council Mitochondrial Biology Unit, Cambridge (UK)
(b) Present address: European Bioinformatics Institute, Cambridge (UK)
References
[1] L.A. Sazanov and P. Hinchliffe, Science 311, 1430-6 (2006).
[2] R.G. Efremov and L.A. Sazanov, Nature 476, 414-20 (2011)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.