- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2006
- Materials Science
- Spatiotemporal reaction kinetics probed by picosecond X-ray diffraction
Spatiotemporal reaction kinetics probed by picosecond X-ray diffraction
Structural dynamics in the solution phase, the environment most relevant to biology and industrial chemistry, are quite complex due to the presence of solvent molecules near solute molecules. Typically solute molecules are dissolved in the ocean of solvent molecules and complex solvation cage structures are formed. Sometimes the photo-reacted atoms or molecules can be trapped inside the cage and recombine with each other (geminate recombination). In another case, the reacted species can escape from the cage and recombine with others from other cages (non-geminate recombination). Historically, time-resolved optical spectroscopy methods have provided ample information about such processes. However, more insight into the dynamics of molecular structure in solution can be obtained using time-resolved X-ray diffraction. It was recently demonstrated that the transient structure in solution can be determined by using time-resolved liquid X-ray diffraction [1].
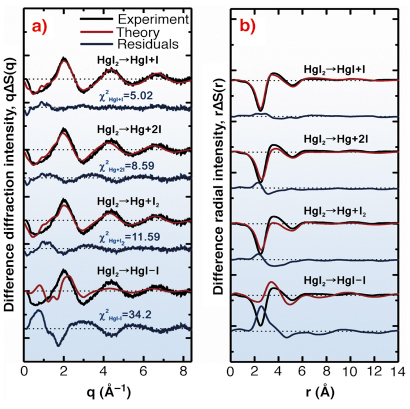 |
|
Fig. 32: Determination of primary photodissociation pathway of HgI2 in methanol solution. The experimental difference curves in q (a) and r space (b) at 100 ps are compared with theoretical curves using four possible putative primary pathways. The channel of two-body dissociation (HgI2 |
In this work, 100 ps time-resolved liquid diffraction experiments on the photoreactions of diiodomercury (HgI2) dissolved in methanol (10 mM) were performed at beamline ID09B, using the optical pump and X-ray probe method. This molecular system has been investigated using the same technique as before, but the transient pathway and subsequent structural dynamics have not been elucidated due to the poor signal-to-noise ratio and limited data analysis method [2]. An optical pulse (267 nm, 2 ps, 986 Hz) triggers impulsive photodissociation of HgI2, followed by subsequent reactions and then the X-ray pulse (100 ps, 0.067 nm, 986 Hz) interrogates the reacting sample by making the diffraction patterns on the CCD detector in a stroboscopic manner for the different time-delays spanning from 100 picosecond to 1 microsecond. The difference diffraction and corresponding radial intensities were extracted from the diffraction pattern and analysed using the so-called “global-fitting method” which considers time-dependent changes of solute and cage structures and changes of solvent structure, itself due to the time-dependent heat and density changes. Among four putative primary reaction channels (Figure 32), a two-body dissociation channel is the dominant dissociation pathway. After this primary bond fission, two parallel recombination processes proceed. Transient intermediate, HgI, associates with an iodine atom to form HgI2 nongeminately, and I2 is formed by nongeminate recombination of two iodine atoms (Figure 33) in several tens of nanoseconds. In the present work, we could clearly show two main conclusions. (1) The determination of transient species in solution is strongly correlated with solvent energetics, which depend on putative reaction channels due to the heat transfer to the solvent. Time-resolved X-ray diffraction can serve as an ultrafast calorimeter in addition to being a sensitive structural probe. (2) A manifold of structural channel can be resolved at the same time with high-precision measurement and global analysis.
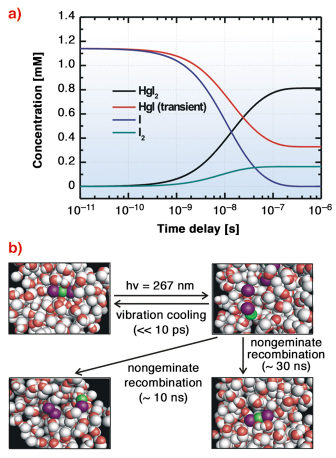 |
|
Fig. 33: Spatiotemporal reaction kinetics determined by time-resolved X-ray diffraction. a) The population changes of chemical species in the photodissociation of HgI2 in methanol solution. b) A schematic reaction mechanism of HgI2 photodissociation in methanol solution. |
Even if there are some limitations, time-resolved X-ray diffraction has matured sufficiently to provide the structure of the intermediates, correlation with solvent environment, and bulk properties of the solvent molecules in solution with unprecedented spatial and time resolution. The success of this work should stimulate future research using such outstanding capabilities for unravelling the structural dynamics of nanomaterials and proteins in the solution phase using time-resolved X-ray diffraction.
References
[1] H. Ihee, M. Lorenc, T.K. Kim, Q. Y. Kong, M. Cammarata, J.H. Lee, S. Bratos, and M. Wulff, Science 309, 1223 (2005).
[2] A. Geis, M. Bouriau, A. Plech, F. Schotte, S. Techert, H.P. Trommsdroff, M. Wulff, and D. Block, J. Luminescence 94, 493 (2001).
Principal Publication and Authors
T.K. Kim (a), M. Lorenc (b), J.H. Lee (a), M. Lo Russo (b), J. Kim (a), M. Cammarata (b), Q.Y. Kong (b), S. Noel (b), A. Plech (c), M. Wulff (b), and H. Ihee (a), Proc. Natl. Acad. Sci. USA 103, 9410 (2006).
(a) Department of Chemistry and School of Molecular Science (BK21), Korea Advanced Institute of Science and Technology (KAIST) (Republic of Korea)
(b) ESRF
(c) Fachbereich Physik der Universität Konstanz (Germany)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.