- Home
- Physisorption vs. chemisorption: adsorption of copper-phthalocyanine on noble metal surfaces
Physisorption vs. chemisorption: adsorption of copper-phthalocyanine on noble metal surfaces
The growth of large π-conjugated molecules on noble metal surfaces has recently attracted much interest in the surface science community. Most studies concentrate on metal-organic interfaces and their geometric and electronic properties since they strongly influence the electronic transport in electronic devices. A very delicate parameter is the interaction strength between the molecules and the substrate because this determines the charge exchange at the metal-organic interface. The normal incidence X-ray standing wave (NIXSW) technique, which could be performed at beamline ID32 until recently, allows the adsorption strength to be quantified by measuring the adsorption height of individual atomic species with an otherwise unreachable precision of < 0.05 Å.
Using low-energy electron diffraction (LEED), ultraviolet photoelectron spectroscopy (UPS), and NIXSW, we have investigated the adsorption of copper-phthalocyanine (CuPc) for different coverages on three substrates: Au(111), Ag(111) and Cu(111) [1-3]. At very low coverages, disordered layers were found in all cases. On increasing coverage or cooling to low temperature (LN2), the disordered layers transform to long-range ordered films. On Au(111) these ordered structures show merely point-on-line registry with the substrate (or are even incommensurate) [1]. UPS data indicate that no charge transfer across the metal-organic interface takes place. On Cu(111) the behaviour is very different [1]. Already above 0.76 monolayers (ML) the molecules are arranged in a commensurate superstructure with one molecule per unit cell, which indicates a favourable adsorption site for the molecules. In UPS a spectral feature just below the Fermi energy is visible and proves charge transfer from the Cu(111) electronic states into the lowest unoccupied molecular orbital (LUMO). CuPc on Ag(111) [2] represents an intermediate case: It forms both point-on-line (at coverages > 0.89 ML) and commensurate superstructures (at low temperature between 0.75 and 0.89 ML), and UPS revealed a half filled LUMO located directly at the Fermi-level. Our XSW study on these three systems aimed at linking these findings with the vertical geometric structure of the interfaces.
 |
|
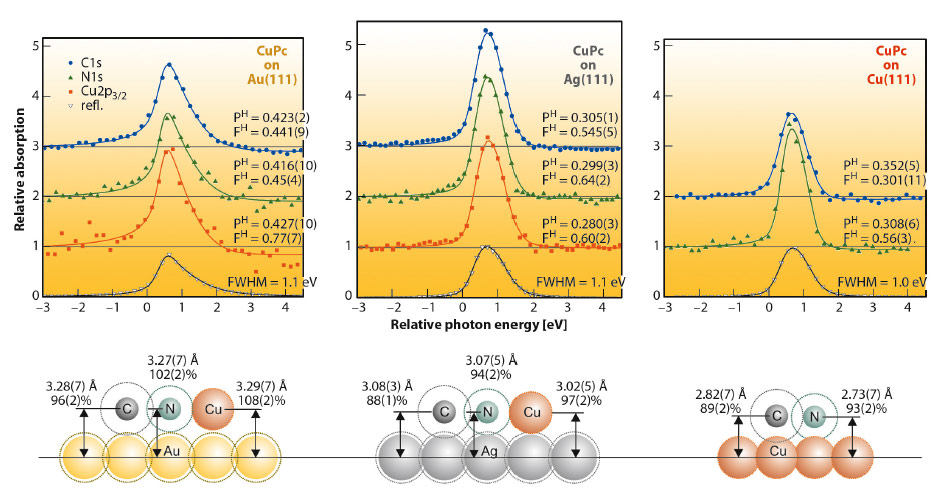
Fig. 131: XSW data (symbols) and fit results (lines) for a closed layer of CuPc on Au(111), Ag(111) and Cu(111) at LN2 temperature. Schematics show the size of all species on scale. Ball diameters represent covalent bonding radii, larger dashed circles correspond to van der Waals radii. Measured bonding distances are given in angstrom and in % of the corresponding van der Waals bonding distances. The height of Cu on Cu(111) could not be obtained due to the bulk-dominated PE signal. |
In Figure 131 NIXSW data and some on-scale schematics of the adsorption heights are shown for all three systems. In an NIXSW scan the photon energy is scanned through a Bragg reflection while the reflected X-ray intensity (grey triangles) and the partial photoelectron yields of all atomic species (coloured symbols) are recorded. Fitting the yield curves results in structural parameters PH (coherent position) and FH (coherent fraction). The first corresponds to the mean adsorption height; the latter reflects the quality of vertical order (FH = 0 corresponds to complete disorder, FH = 1.0 to perfect order).
The differences in the adsorption heights for the three systems (≈ 3.3 Å, ≈ 3.05 Å and ≈ 2.8 Å for CuPc/Au(111), Ag(111) and Cu(111), respectively) clearly indicate an increasing interaction strength for this row. However, when comparing these absolute numbers, one neglects the differences in the size of the individual atomic species. Since Cu is significantly smaller than Au and Ag, one should rather consider bonding distances normalised by van der Waals radii. These “relative bonding distances” indicate physisorption of the molecules on Au(111) (values around 100%), but chemisorption on Ag(111) and Cu(111) (88–94% for C and N). The schematics in Figure 131 illustrate the situation: Van der Waals radii of all species (dotted circles) overlap in the case of CuPc on Cu(111) or Ag(111), but not on Au(111).
These findings are in excellent agreement with UPS results. Chemisorptive interaction for CuPc on Ag(111) and Cu(111) and physisorption for CuPc on Au(111) are both compatible with the charge transfer found in electron spectroscopy. It also explains the differences in the lateral structure formation for the adsorbate layers: In the case of a strong molecule-substrate interaction (chemisorption), the substrate dictates the structure formation in the organic layer and enforces a commensurate registry. In the case of weaker, physisorptive interaction, the overlayer structure is dominated by the intermolecular interaction and hardly influenced by the substrate [3]. Point-on-line or incommensurate structures are the consequence. In summary, we could reveal this delicate interplay of (vertical) molecule-substrate and (horizontal) intermolecular interactions and identify the driving force for the lateral structure formation.
Principal publication and authors
I. Kröger (a), B. Stadtmüller (a), C. Kleimann (a), P. Rajput (b) and C. Kumpf (a), Phys. Rev. B 83, 195414 (2011).
(a) Peter Grünberg Institut (PGI-3), Forschungszentrum Jülich, and Jülich-Aachen Research Alliance (JARA) – Fundamentals of Future Information Technology (Germany)
(b) ESRF
References
[1] B. Stadtmüller, I. Kröger, F. Reinert and C. Kumpf, Phys. Rev. B 83, 085416 (2011).
[2] I. Kröger, B. Stadtmüller, C. Stadler, J. Ziroff, M. Kochler, A. Stahl, F. Pollinger, T.L. Lee, J. Zegenhagen, F. Reinert and C. Kumpf, New. J. Phys. 12, 083038 (2010).
[3] I. Kröger, B. Stadtmüller, C. Wagner, C. Weiss, R. Temirov, F.S. Tautz and C. Kumpf, J. Chem. Phys. 135, 234703 (2011).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.