- Home
- Structure of the torque ring of the bacterial flagellar motor and the molecular basis for rotational switching
Structure of the torque ring of the bacterial flagellar motor and the molecular basis for rotational switching
The bacterial flagellar motor is one of the most efficient rotary motors known to man [1]. It utilises proton motive force to drive the rotation of long helical flagellar filaments at up to 1000 revolutions per minute, yet it can reverse its direction in milliseconds. The motor and filament act as a propeller for swimming bacteria, which move in cycles of relatively lengthy forward runs, followed by short tumbles that alter the trajectory of the cell. These cycles are essential for the survival of bacteria as they bias their runs and tumbles such that the net movement is towards a favourable environment and away from unfavourable ones. The mode of swimming is determined by the direction of rotation of the flagellar motor. A run occurs during counter-clockwise (CCW) rotation when all filaments form a helical bundle that propels the cell forward. A tumble occurs when at least some flagella rotate in a clockwise (CW) direction, which causes the bundle to disassemble and results in a somersaulting of the cell. Similar to ATP synthases, the bacterial flagellar motor uses the potential energy from an electrochemical gradient of cations across the cytoplasmic membrane to generate torque. Remarkably, in the flagellar motor both CCW and CW rotation are driven by a flow of cations towards the cytosol, whereas in the reversible bacterial ATP synthases, a switch of rotation is connected to a reversal of cation flow.
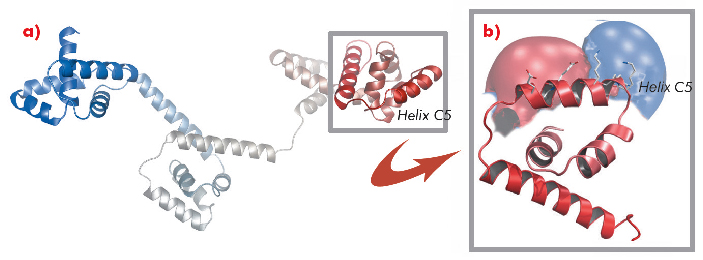
A protein known as FliG forms a ring in the motor that is involved in rotational switching and interacts with the cation channel forming stator subunit MotA to generate torque. We determined the crystal structure of the full-length FliG protein from the hyperthermophilic bacterium Aquifex aeolicus to 2.4 Å resolution using native data collected at beamline ID14-1 of the ESRF coupled with single anomalous dispersion data sets from crystals containing selenomethionine-substituted protein collected at beamline 14-ID-B of the Advanced Photon Source (Figure 113).
 |
|
Fig. 113: a) Structure of the full-length A. aeolicus FliG monomer. The N-terminus is shown in blue, the C-terminus in red. b) The C-terminal domain containing torque helix C5 is shown enlarged and an electrostatic surface potential map is superimposed to demonstrate the clear distribution of charge along the helix. |
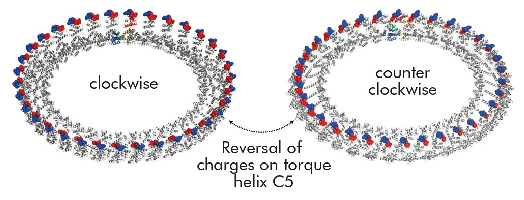
Structural differences to a previously solved crystal structure of the middle and C-terminal domains of FliG from Thermotoga maritima [2] correlated strongly to the location of dozens of mutations that bias the direction in which the motor rotates and revealed two striking conformational changes that are involved in rotational switching. Furthermore, conserved crystal contacts revealed how armadillo repeat motifs on adjacent FliG monomers interact to form a right-handed superhelix and hence define the structural basis for an intermolecular FliG-FliG interaction that mediates the assembly of the FliG ring. This allowed us to derive a model of the FliG ring that is consistent with the size, shape and symmetry of 3D electron microscopy reconstructions and with intermolecular contacts that were defined by mutagenesis and cross-linking studies. Finally, in the context of a fully assembled ring, conformational changes in FliG redistribute the charges involved in torque generation providing a compelling model for rotational switching (Figure 114).
 |
|
Fig. 114: Model of the FliG ring in CW and CCW conformations. Note that the distinct charge distribution on torque helix C5 is reversed. |
Principal publication and authors
L.K. Lee (a), M.A. Ginsburg (a), C. Crovace (b), M. Donohoe (a) and D. Stock (a), Nature 466, 996-1000 (2010).
(a) Structural and Computational Biology Division, Victor Chang Cardiac Research Institute, Sydney (Australia).
(b) MRC Laboratory of Molecular Biology, Cambridge (UK)
References
[1] T. Minamino, K. Imada and K. Namba, Curr. Opin. Struct. Biol. 18, 693-701 (2008).
[2] P.N. Brown, C.P. Hill and D.F Blair, EMBO J. 21, 3225-3234 (2002).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.