- Home
- Molecular basis of the alternating access model of membrane transport in a sodium-hydantoin transporter
Molecular basis of the alternating access model of membrane transport in a sodium-hydantoin transporter
Secondary active membrane transport proteins harness electrochemical gradients to facilitate the movement of small molecules or ions across membranes. They are proposed to use an alternating access model of transport [1], the principle of which is that the protein switches conformations to present the substrate binding site to alternate sides of the membrane without ever fully opening a channel from one side to the other. Due to the difficulty of elucidating the structures of these relatively unstable proteins in multiple states there has been a paucity of molecular detail regarding the mechanism.
 |
|
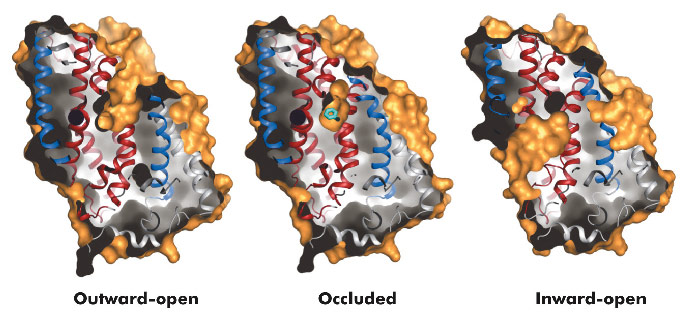
Fig. 96: Surface representations of the three conformational states of Mhp1. |
The hydantoin transporter from Microbacterium liquefaciens, Mhp1, belongs to the nucleobase cation symport-1 family responsible for transporting nucleobases into bacteria, fungi or plants. The uptake of hydantoins by Mhp1 is driven by the concentration gradient of sodium between the two sides of the membrane, probably with one sodium ion being transported for every hydantoin substrate. Sodium is present at a lower concentration inside the bacteria relative to the surrounding environment. Mhp1 is comprised of 12 transmembrane helices [2] with a fold that appears to be widespread amongst secondary active transporters [3]. Previously we reported the structures of Mhp1 in two different conformations, an outward-open form with sodium bound and an occluded form with both sodium and substrate present. We have now solved the crystal structure of an inward-open form (Figure 96). Data extending to a resolution of 3.8 Å were collected on ID29. The structure was solved by the selenium SAD method following location of the selenium atoms using initial phases from molecular replacement and density modification. The refined structure clearly shows the substrate-binding site to be open on the inward-facing side and sealed completely on the outside (Figure 96).
 |
|
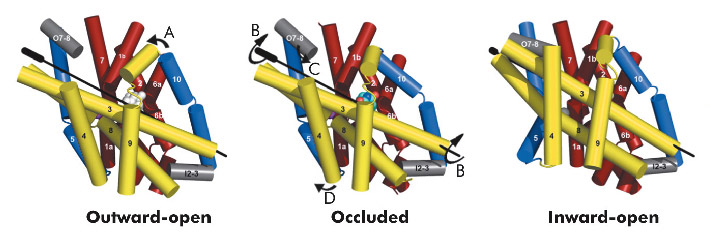
Fig. 97: Conformational changes between the three states of Mhp1. The hash motif is coloured yellow, the four helix bundle red and the flexible helices blue. The C-terminus and loops have been omitted for clarity. The arrows show the movements. A) A flexible helix bends over the substrate; B) The hash motif rotates around the axis shown. C) A small extracellular helix (shown in grey) packs onto the outside of the membrane; D) A second flexible helix bends to open the pocket on the inside. |
Combining this crystal structure with those we have previously determined, we have been able to postulate how the protein can move from one state to the other. In this regard, it is convenient to consider Mhp1 as being composed of the following units: a four helix bundle, a “hash motif” so called because the four helices in this motif resemble the # sign, two “flexible helices” and the C-terminal region (Figure 97). The sodium and substrate binding sites are located at the interface of the bundle and hash motifs. Upon substrate binding in a large cavity that can be seen in the outward open structure, one of the flexible helices bends to block the substrate in its binding site thus occluding it from the outside. The protein can then undergo a more dramatic conformational change to the inward-facing state. The major difference between the outward and inward-facing structures is that the hash motif has rotated 30° relative to the bundle. This movement effectively opens the substrate binding site on the inward-facing side and further blocks it to the exterior. This movement is accompanied by some smaller changes as shown in Figure 97. The rotation of the hash motif relative to the bundle effectively destroys both the sodium and substrate binding sites. The binding of sodium therefore will pull the equilibrium between the inward and outward-facing states towards the latter. In this conformation the protein is primed to bind the substrate. Upon binding the substrate the outward-facing state will be destabilised allowing the protein to open up to the cytoplasm where the sodium and substrate can diffuse into the bacteria.
Principal publication and authors
T. Shimamura (a,b,c), S. Weyand (a,b,d), O. Beckstein (e), N.G. Rutherford (f), J.M. Hadden (f), D. Sharples (f), M.S.P. Sansom (e), S. Iwata (a,b,c,d,g), P.J.F. Henderson (f) and A.D. Cameron (a,b,d), Science 328, 470-473 (2010).
(a) Division of Molecular Biosciences, Membrane Protein Crystallography Group, Imperial College, London (UK)
(b) Human Receptor Crystallography Project, ERATO, Japan Science and Technology Agency, Kyoto (Japan)
(c) Department of Cell Biology, Graduate School of Medicine, Kyoto University (Japan)
(d) Membrane Protein Laboratory, Diamond Light Source (UK)
(e) Department of Biochemistry, University of Oxford (UK)
(f) Astbury Centre for Structural Molecular Biology, Institute for Membrane and Systems Biology, University of Leeds (UK)
(g) Systems and Structural Biology Center, RIKEN (Japan)
References
[1] O. Jardetzky, Nature 211, 969-970 (1966).
[2] S. Weyand et al., Science 322, 709-713 (2008).
[3] J. Abramson and E. Wright, Curr Opin Struct Biol 19, 425-32 (2009).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.