- Home
- Subnanosecond-resolved Diffraction: A New Dimension for the Study of Atomic Positions in Moving Molecules
Subnanosecond-resolved Diffraction: A New Dimension for the Study of Atomic Positions in Moving Molecules
Introduction by M. Wulff, ESRF
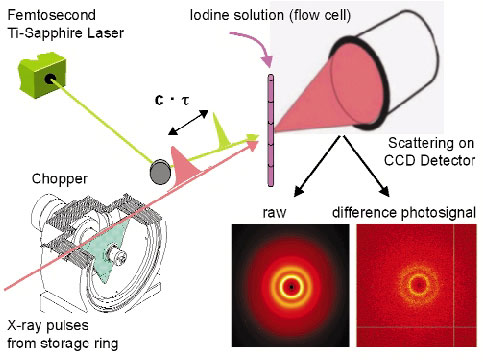
It is now possible to use the pulsed X-ray beam to "film" the structure of a laser-excited molecules as they evolve though internal conversion or interaction with the environment. A specialised setup, sometimes called the molecular camera, has been built on ID09B for this purpose. The camera can record the transient structure of liquids, glasses and crystals by time-resolved diffraction to a time resolution of 100 picoseconds, a limit set by the X-ray pulse length. The main components in the camera are an X-ray chopper, a femtosecond laser and a CCD detector (Figure 59). The experiments become time resolved by controlling the delay between the laser and X-ray pulse. The camera is used for Laue diffraction from proteins, for small molecule and powder diffraction and for angular dispersive diffraction from glasses and liquids. This new ultrafast X-ray diffraction technique (UXD) is complementary to ultrafast electron diffraction (UED) since the latter is limited to gas-phase samples due to the high scattering power of electrons.
 |
|
Fig. 59: The molecular camera for picosecond diffraction experiments. |
The most striking films are taken with excited protein crystals such as the carbon monoxide complex of myoglobin and hemoglobin. In the following article by F. Schotte et al., they show how myoglobin directs the dissociated CO away from its initial binding site next to iron. The CO migrates though a network of tiny cavities, to finally end up in the solvent outside of the protein. The ability of the protein to shield the CO from direct recombination is an intricate feature of myoglobin, which is essential for respiration. The quality of the data depends on the ability to dissociate, non-destructively, a substantial fraction of unit cells. It turned out to be beneficial to increase the laser pulse length from 150 fs to 2 ps to maximise the dissociation yield. This trick increased the number of "correctly excited" unit cells by 10, which gave a 10-fold improvement in the signal to noise. This effect is due to the presence of short-lived, high-absorption electronic states that tend to obstruct the entry of the laser pulse.
The experiments on excited molecules in liquids are reported subsequently by A. Plech et al., who studied the excited-state structure of iodine in liquid CCl4. The initial aim was to extract the diffracted signal from excited iodine, determine the change in the I2 bond length and the degree of dissociation. It came as a surprise, therefore, to see that a local change in structure around iodine can induce a global change in structure in the bulk liquid, driven by the flow of energy from the solute to the liquid. This forces the liquid to expand. The expansion shows up after 10 ns and is over after 100 ns. This pattern is quite general in photo-excited liquids and we now classify the liquid structure into a pre-expansion, an expansion and post-expansion phase. The expansion amplitude, defined by the change in distance between neighbouring solvent molecules, is typically a fraction of a milliangstrom, but is shows up since it integrates all the solvent molecules in the sample. From the expansion amplitude, it is possible to check the associated energy input from the laser, which is a way to check if the total energy is conserved.
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.