- Home
- Dispersive EXAFS Studies of Rapid, Adsorbate-induced, Phase Change in Rh Catalysts and Its Impact on the Selective Reduction of NO to N2 by H2
Dispersive EXAFS Studies of Rapid, Adsorbate-induced, Phase Change in Rh Catalysts and Its Impact on the Selective Reduction of NO to N2 by H2
Dispersive EXAFS Studies of Rapid, Adsorbate-induced, Phase Change in Rh Catalysts and Its Impact on the Selective Reduction of NO to N2 by Hb2
Rhodium is a principal component in modern auto exhaust catalysts due to its capacity to convert the environmentally undesirable NO to benign N2. Therefore, the fundamental chemistry of Rh in this respect has been much studied [1]. These studies have resulted in the following conundrum: models based on single crystal Rh samples fail to reproduce the activity and selectivity of a highly-dispersed Rh catalyst in fact the latter consistently produces significant levels of N2O, whereas this is never observed from the ideal Rh surfaces [1]. This contradictory behaviour points to a major gap in our understanding of these systems and questions the often made assumption that metallic single crystals can be regarded as valid models of their highly-dispersed counterparts.
By using the Laue monochromator configuration [2] available at beamline ID24, we have used time-resolved energy dispersive EXAFS (EDE) at the Rh K-edge to probe the dynamic behaviour of supported Rh catalysts under feedstocks containing NO.
 |
|
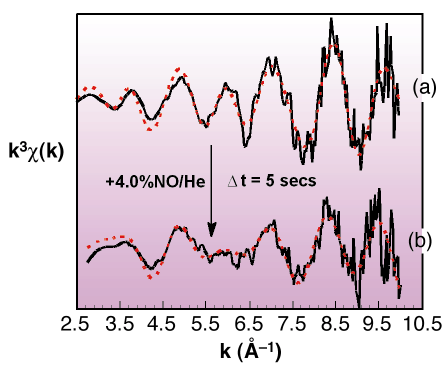
Fig. 45: Rh K-edge derived k3-weighted EXAFS data from (a) a reduced 4wt% Rh/Al2O3 sample and (b) after exposure to 4% NO/He for ca. 5s. Black and red lines indicate experimental data and theoretical fits respectively. |
Figure 45 shows an example of how the metallic Rh nanoparticles structure responds to exposure to NO. Switching from a gas feed of He to one of 4% NO in He resulted in an instantaneous change in the Rh EXAFS signal consistent with a transition from metallic to a new oxidic phase resembling Rh2O3. This observation was accompanied by a considerable exotherm. This, together with simultaneously obtained mass spectrometer data, shows that in the nano-regime, molecular dissociation, and the concomitant release of energy, can effect rapid and extreme changes in the structure of the supported phase: no comparable analogy of this behaviour exists on extended metal surfaces.
This observation has a further logical consequence: if a metallic Rh phase is ever to be attained in a reactive environment containing such oxidants (as in a car exhaust with an engine running under "lean burn" conditions), it may only do so as a result of a dynamic equilibrium between these oxidative processes and reactions that may reverse them.
 |
|
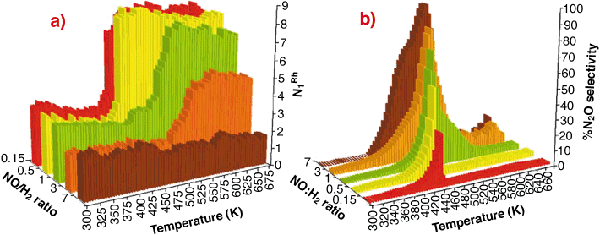
Fig. 46: (a) Variation in N1Rh derived from analysis of EXAFS spectra collected during NO reduction by H2 over of 5wt% Rh/ |
 -Al2O3 catalysts as a function of feedstock composition and reaction temperature; (b) Selectivity toward N2O production as a function of reaction temperature and active feedstock composition in the reduction of NO by H2 over 5wt% Rh/
-Al2O3 catalysts as a function of feedstock composition and reaction temperature; (b) Selectivity toward N2O production as a function of reaction temperature and active feedstock composition in the reduction of NO by H2 over 5wt% Rh/Figure 46ashows the variation in first shell Rh-Rh co-ordination (N1Rh) observed during reaction with NO/H2/He feedstocks of varying composition (7 > NO: H2 > 0.15) as a function of temperature. In these cases N1Rh = 2 corresponds to the fully formed oxidic phase, N1Rh = 7-8 indicating fully reduced metallic Rh particles (ca. 20 Å diameter). Figure 46b shows the selectivity to N2O derived simultaneously with the EXAFS data.
This clearly shows that the basic chemistry outlined in Figure 45 has a pervasive influence on the behaviour of the catalyst. It is only under the most reducing conditions (NO/H2 < 1) that a "metallic" Rh phase is ever observed. Moreover, the levels of N2O produced can be directly correlated with the presence of highly-oxidised Rh, and especially with the collapse of this phase to yield a metallic Rh phase. However when metallic Rh is formed, we see that it produces practically no N2O in keeping with the behaviour expected from that of Rh single crystals.
In summary, the application of time-resolved energy-dispersive EXAFS/mass spectrometry has allowed us to show that N2O production in supported Rh systems results predominantly from an oxidised phase of Rh and its eventual collapse to form metallic Rh. That such an oxidised phase is formed at all is a consequence of an intrinsically enhanced reactivity of nanoscale Rh towards NO a behaviour that is unique to nanoparticulate Rh and that therefore cannot be modelled through the use of systems of intrinsically lower dispersion.
References
[1] For instance, V.P. Zhdanov and B. Kasemo, Surf. Sci. Rep. 29, 31 (1997).
[2] M. Hagelstein, C. Ferraro, U. Hatje, T. Ressler and W. Metz, J. Synchrotron Rad. 5, 1396 (1998).
Principal publication and Authors
M.A. Newton (a), A.J. Dent (b), S. Diaz-Moreno (c), S.G. Fiddy (c) and J. Evans (a), Angewandte Chem. Intl. Ed. 41, 2587 (2002); T. Campbell (a), A.J. Dent (b) , S. Diaz-Moreno (c), J. Evans (a), S.G. Fiddy (c), M.A. Newton (a) and S. Turin (a), Chem. Comm., 304 (2002).
(a) University of Southampton (UK)
(b) SRS Daresbury (UK)
(c) ESRF
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.