- Home
- Fast Time-resolved in situ Powder-diffraction Studies of High-temperature Oxidation/Reduction Reactions
Fast Time-resolved in situ Powder-diffraction Studies of High-temperature Oxidation/Reduction Reactions
In order to perform fast time-resolved in situ powder diffraction experiments of chemical reactions, a rotating-slit system for the MAR345 imaging-plate system has been developed for BM01B (SNBL) (Figure 126). A screen with a wedge-shaped opening is rotated in front of the MAR345 image-plate detector. The time resolution can be adjusted by varying the slit size and the rotation speed. In situ powder diffraction data have been collected with a time resolution around 100 ms.
 |
Fig. 126: The rotating slit system at the MAR345 diffractometer at SNBL. A capillary-based in situ micro reaction cell and a hot air heater are mounted on the diffractometer (inset). |
The rotating-slit system has been used in time-resolved in situ powder diffraction studies of the fast oxidation/reduction reactions of oxygen ion conducting perovskite type materials at high temperature, 400-800°C.
Perovskite type oxides with composition (A1-xA'x)BO3±![]() , (A = La, Y; A' = Sr, Ca; B = Mn, Fe, Co) are of interest for applications such as catalysis, oxygen permeable membranes, solid oxide fuel cells and colossal magneto-resistance. For these materials, catalytic activity, magnetism and oxygen ion conductivity, are closely related to oxygen stoichiometry, crystal structure and redox properties. The oxidation/reduction reactions are of topotactic nature and are completed within a few seconds at 800°C. Therefore, very fast data collection is required. High-quality powder diffraction data must be collected in order to monitor structural changes during the reaction, preferable by means of Rietveld analysis.
, (A = La, Y; A' = Sr, Ca; B = Mn, Fe, Co) are of interest for applications such as catalysis, oxygen permeable membranes, solid oxide fuel cells and colossal magneto-resistance. For these materials, catalytic activity, magnetism and oxygen ion conductivity, are closely related to oxygen stoichiometry, crystal structure and redox properties. The oxidation/reduction reactions are of topotactic nature and are completed within a few seconds at 800°C. Therefore, very fast data collection is required. High-quality powder diffraction data must be collected in order to monitor structural changes during the reaction, preferable by means of Rietveld analysis.
A capillary based micro reaction cell, allowing a flow of gas to pass through the sample, was used for the experiments [1,2]. Using a remote-controlled three-way valve, abrupt changes between nitrogen and oxygen gas can be achieved, allowing kinetic information to be extracted from variation of intensity or position of selected Bragg reflections. Samples were contained in 0.7-1mm quartz glass capillaries mounted in a Swagelok fitting. The sample was heated using a hot air blower.
Results are presented for oxidation of SrFe0.97Cr0.03O2.6 at 700-800°C. Switching from a nitrogen to an oxygen atmosphere is done while the slit is rotating continuously in front of the imaging plate (one rotation in 15 seconds). Figure 127 shows the changes in one of the reflections as the oxidation proceeds at 800°C. The gas flow was switched from N2 to O2 at time t = 0, and powder diffraction data were integrated to provide a time resolution of 100 ms. An almost instantaneous change in the diffraction pattern was observed.
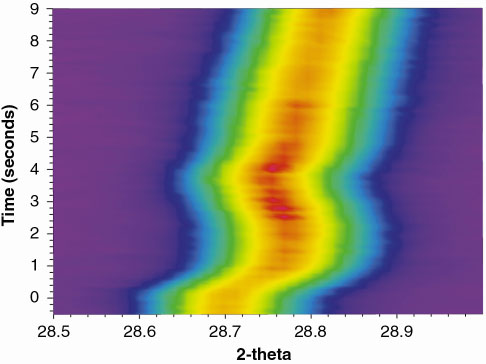 |
Fig. 127: Changes in position and intensity of one diffraction peak during oxidation of SrFe0.97Cr0.03O3- |
The unit cell volume normally decreases during the oxidation of perovskite oxides. This is caused by shortening of the metal-oxygen bond on increased valence state of the cation, together with the efficient packing of coordination polyhedra. As an example, consider the oxidation of SrFeO2.6:
SrFe(III)0.8Fe(IV)0.2O2.6 + 0.1O2 ![]() SrFe(III)0.4Fe(IV)0.6O2.8
SrFe(III)0.4Fe(IV)0.6O2.8
Typically the unit cell decrease mirrors the variation in oxygen stoechiometry. For the present oxidation process, the time-resolved data reveal an unexpected increase in the pseudo-cubic unit cell parameter (using LeBail profile refinement) for an intermediate time interval (Figure 128). It can be clearly seen that the oxidation process proceeds in two steps. After an initial fast decrease, a plateau is reached, most visible at 775°C. A maximum in the unit cell volume is encountered before the final (slower) decrease occurs. The same general features are observed at 700°C, where the process is much slower.
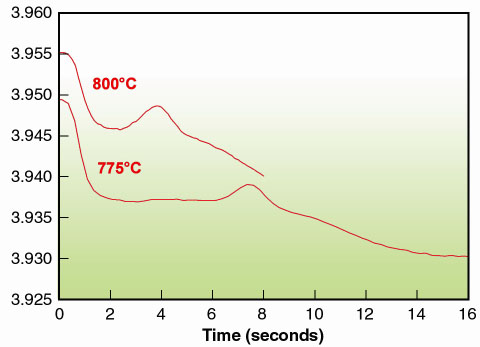 |
Fig. 128: Unit cell parameter (pseudo cubic) of SrFe0.97Cr0.03O3- |
So far thermogravimetric experiments have not shown mass variations indicating a two-step oxidation process. The reactions are too fast for in situ neutron powder diffraction studies that otherwise could determine the time evolution of the atomic coordinates for the light oxygen atoms. It is presently proposed that the expansion anomaly is caused by redistribution of oxygen vacancies, thereby changing the relative amount of lower coordinated MO4 tetrahedra and MO5 square pyramids. Forthcoming in situ synchrotron X-ray diffraction experiments will hopefully solve this intriguing problem.
References
[1] P. Norby, J. Amer. Chem. Soc., 119, 5215-5221 (1997).
[2] E. Krogh Andersen, I.G. Krogh Andersen, P. Norby and J.C. Hanson J. Solid St. Chem. 141 235-240 (1998).
Principal Publication and Authors
P. Norby (a), H. Fjellvåg (a) and H. Emerich (b), in preparation.
(a) Department of Chemistry, University of Oslo (Norway)
(b) SNBL, ESRF
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.