- Home
- News
- Spotlight on Science
- First structure...
First structure and inhibitor of the emerging cancer target MTHFD2
17-02-2017
Mitochondrial MTHFD2 is highly upregulated in cancer cells to support purine and thymidylate synthesis. MTHFD2 is normally only expressed during embryonic development and not in adult cells. The structure of MTHFD2 and identification of an inhibitor now provides a promising foundation for the development of potent cancer drugs.
Rapidly dividing cells depend on one-carbon units that are supplied for synthesis of several important compounds in the cell, most importantly the purine bases adenine and guanine, and the pyrimidine base thymidine. The one-carbon units are attached to tetrahydrofolate (THF) molecules oxidised to different states depending on application. One of the enzymes facilitating these oxidations is the mitochondrial methylene-THF dehydrogenase and cyclohydrolase (MTHFD2), which has previously been found to be highly expressed in several types of cancer cells. Normally expressed in embryonic cells and not in adult cells, MTHFD2 is a very interesting target for cancer treatment due to the possibility to kill cancer cells without harm to normal cells. The first structure of MTHFD2 together with the first inhibitor for MTHFD2 is now giving us the opportunity to develop potent and selective inhibitors.
First discovered in Ehrlich ascites tumour cells in 1960 and later described as a mitochondrial NAD+-dependent methylene-THF dehydrogenase and cyclohydrolase expressed in embryonic and transformed cells, MTHFD2 needs both NAD+ and inorganic phosphate as cofactors to function. In the same protein family, the use of NADP+ has also been found, for example, in the cytosolic trifunctional MTHFD1 that is part of the same enzyme family as MTHFD2 and shares the dehydrogenase and cyclohydrolase functions. MTHFD1 also carries out a 10-formyl-THF-synthetase function in a second domain missing in MTHFD2.
LY345899 is a substrate-based inhibitor originally developed for MTHFD1 and very closely resembles the methylene-THF that is a substrate for both MTHFD1 and MTHFD2. To show target engagement of the inhibitor against MTHFD2, we performed several in vitro and in vivo assays. An in vitro dose-response curve for both MTHFD2 and MTHFD1 determined IC50 values for the inhibition, 663 nmol/L for MTHFD2 and 96 nmol/L for MTHFD1. Binding of LY345899 to MTHFD2 was confirmed by differential scanning calorimetry (DSF), where addition of LY345899 increased the protein melting point from 44°C to 55°C. To confirm binding in a cellular context, cellular thermal shift assays (CETSA) and drug affinity responsive target stability assays (DARTS) were also performed. CETSA show an increased stability of the protein in cell lysates with the inhibitor added. DARTS show an increased stability of MTHFD2 against protease degradation when incubated with inhibitor in cell lysates. As for CETSA, a similar effect is not seen when using intact cells. Both CETSA and DARTS measure protein stability in a cellular context upon addition of the inhibitor and in both cases MTHFD2 is stabilised by LY345899, a clear sign of target engagement.
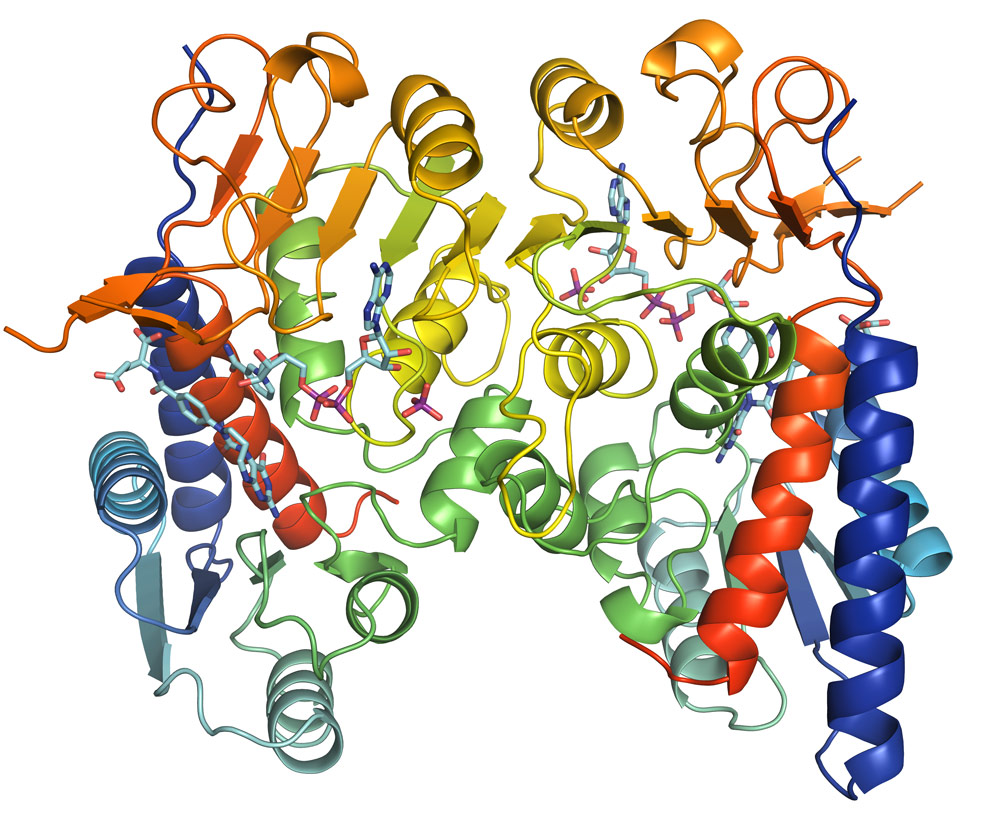
Following data collection at beamline ID30A-3, the crystal structure was determined to a resolution of 1.9 Å. The structure of MTHFD2 shows clearly the binding of both cofactors as well as the inhibitor LY345899 (Figure 1). All substrates bind in a long and wide cleft formed between the N-terminal and C-terminal domain of MTHFD2. The protein is a dimer, as is clearly seen in Figure 1, and an interesting feature is that both monomers interact with inorganic phosphate (Figure 2a).
 |
|
Figure 1. Overall structure of MTHFD2. Monomers are shown as cartoon in rainbow colouring. Ligands (NAD+, LY345899 and inorganic phosphate) for both monomers are shown as sticks (cyan, phosphor coloured purple). |
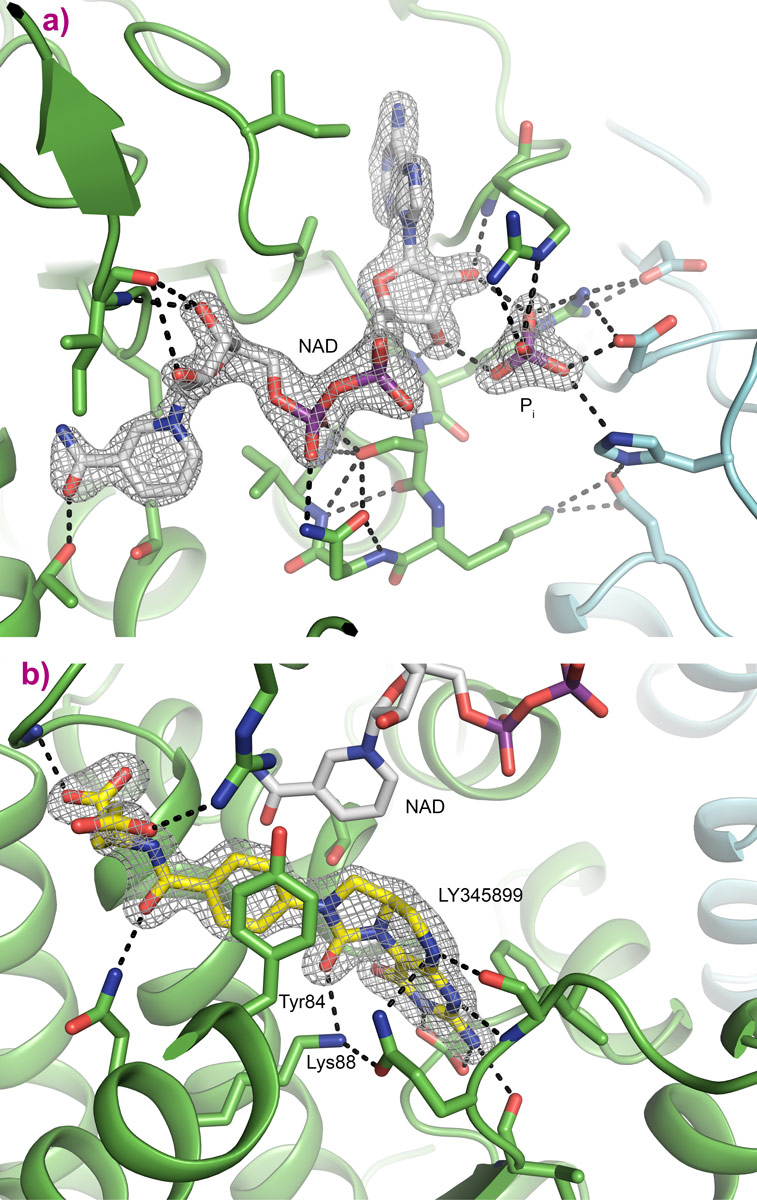
Detailed binding of LY345899 in the active site of MTHFD2 can be seen in Figure 2b, where several hydrogen bonds as well as bridging water molecules help anchor the inhibitor into place. Electron density for the inhibitor is clear and well defined. The YxxxK motif that has been proposed for the binding and cyclohydrolase activity of MTHFD2 is found close to the inhibitor. The nicotinamide moiety of NAD is ideally positioned for the dehydrogenase reaction. The binding of NAD and inorganic phosphate can be seen as an NADP mimicking arrangement even though the protein prefers NAD + phosphate over NADP, Figure 2a.
 |
|
Figure 2. Cofactor and inhibitor binding sites. Electron density: 2Fo-Fc map at 1.5 σ. Monomers shown in green and cyan. Important residues for hydrophobic interactions and hydrogen bonding are shown as sticks. Waters are omitted for clarity. Phosphor is coloured purple. a) Binding of NAD+ (grey) and inorganic phosphate, b) Binding of LY345899 (yellow), with NAD+ shown in grey. |
Evidence of MTHFD2 upregulation and over-expression in cancer cells is clear. The protein is expressed in embryonic cells and transformed cells independent of their tissue of origin, but importantly not in adult healthy cells. The cancer-specific expression of MTHFD2 holds the promise of fewer adverse side-effects compared to many drugs currently used in cancer therapy. Collectively, these aspects make the MTHFD2 protein highly promising as an anticancer target. The crystal structure of MTHFD2 and the identification of the first MTHFD2 inhibitor can now provide a rationale for developing potent and selective MTHFD2 inhibitors.
Principal publication and authors
Crystal structure of the emerging cancer target MTHFD2 in complex with a substrate-based inhibitor, R. Gustafsson (a), A.-S. Jemth (b), N.M.S. Gustafsson (b), K. Färnegårdh (c), O. Loseva (b), E. Wiita (b), N. Bonagas (b), L. Dahllund (d), S. Llona-Minguez (b), M. Häggblad (e), M. Henriksson (b), Y. Andersson (d), E. Homan (b), T. Helleday (b), P. Stenmark (a), Cancer Research 77, 1-12 (2017); doi: 10.1158/0008-5472.CAN-16-1476.
(a) Department of Biochemistry and Biophysics, Stockholm University, Stockholm (Sweden)
(b) Science for Life Laboratory, Division of Translational Medicine and Chemical Biology, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm (Sweden)
(c) Drug Discovery and Development Platform, Science for Life Laboratory, Department of Organic Chemistry, Stockholm University, Solna (Sweden)
(d) Drug Discovery and Development Platform, Science for Life Laboratory, School of Biotechnology, Royal Institute of Technology, Solna (Sweden)
(e) Biochemical and Cellular Screening, Science for Life Laboratory, Department of Biochemistry and Biophysics, Stockholm University, Stockholm (Sweden)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.