- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2013
- Soft condensed matter
- Filming the birth of molecules and solvent rearrangement
Filming the birth of molecules and solvent rearrangement
Solvent plays an important role in solution-phase chemical reactions by serving as an energy source to activate the reaction as well as a heat bath to stabilise the products. As a result, the properties of the solvent significantly affect the energy landscape, rates, and pathways of a reaction in solution. The interplay of solute and solvent molecules and its effect on the outcome of chemical reactions has been a topic of intense research in the field of reaction dynamics over several decades, and the kinetic and spectral signatures of the solvation process have been elucidated on femto- to picosecond time scales [1, 2]. It is still challenging experimentally to probe the bonds that are formed during a chemical reaction, especially the changes between solute and solvent accompanying the structural change of reacting solute molecules.
 |
|
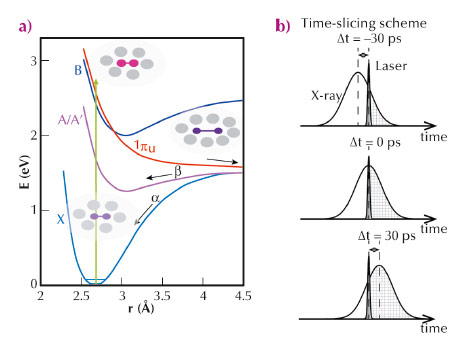
Fig. 89: a) Potential energy surface of I2 in CCl4. The processes α and β represent the geminate recombination of two I atoms in the X and A/A’ states, respectively. The process γ represents nongeminate recombination through the solvent. b) Schematic of the time-slicing experiment. At a negative time delay (for example –30 ps), the X-ray pulse arrives (effectively) earlier than the laser pulse, but the X-ray pulse, which is much longer in time than the laser pulse, is still present after the interaction with the laser pulse and thus scattered off the laser-illuminated sample. At time zero, half of the X-ray pulse probes the laser-illuminated sample. |
Geminate recombination of iodine atoms (Figure 89a) to form molecular I2 in solution after photodissociation is a good example of a prototype solution-phase reaction and has been investigated by spectroscopic studies and quantum chemistry [3]. However, the change in the molecular structure (i.e. bond length change) and the response of the surrounding solvent cage have never been directly observed. Time-resolved X-ray liquidography (solution scattering) is well suited for monitoring this solution-phase reaction because it directly probes the atom-atom distance distribution as a function of time. By using the time-slicing scheme where data are collected at earlier time delays and with finer time increments (down to 10 ps) than the X-ray pulse width (~100 ps) (Figure 89b) and the deconvolution data processing, we can extract the dynamics that occur faster than the X-ray pulse width and monitor the evolution of both the atom-atom distance distribution of iodine atoms in solvent and the solute-solvent distance distribution at the early stages of I-I bond formation within the solvation shell.
 |
|
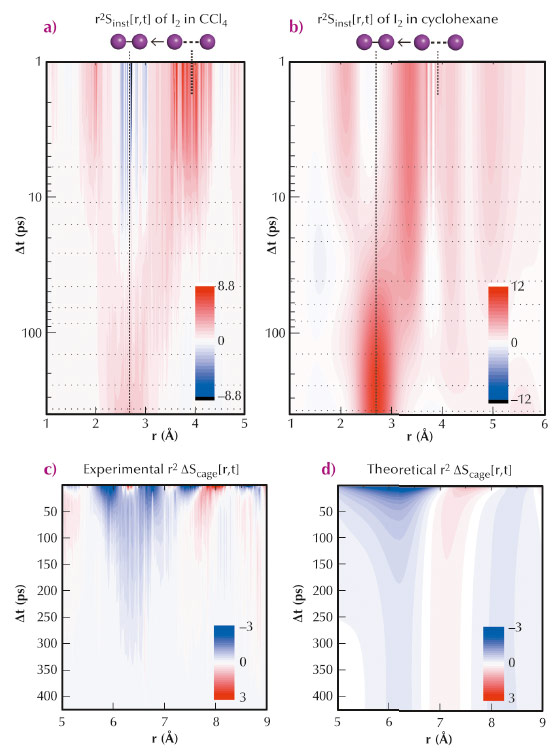
Fig. 90: a) Time-dependent I-I distance distribution functions (r2Sinst[r,t]) corresponding to I2 in CCl4. b) The r2Sinst[r,t] of I2 in cyclohexane. c) The experimental r2∆Sinst[r,t] curves at large r values: time-dependent solute-solvent distance distribution functions. d) Theoretical r2∆Sinst[r,t] based on the experimental data from (a) and MD simulation. |
In this study, we measured the dynamics of geminate recombination and vibrational relaxation of I2 in two different solvents, CCl4 and cyclohexane, in real time using picosecond X-ray liquidography at beamline ID09TR. The birth and vibrational relaxation of I2 molecules and the associated rearrangement of solvent molecules are mapped out in the form of temporally varying interatomic distance distribution. The atom-atom distance distribution clearly shows the time-dependent progression of the I-I distance (Figure 90a,b). The relaxation of elongated iodine distribution is followed by bi-exponentials, which is well in agreement with previous spectroscopic observations [4] and our molecular dynamics (MD) simulation. Also iodine atoms can be separated by larger distances in cyclohexane than in CCl4 and the vibrational relaxation of a newly born “hot” I2 molecule occurs faster in CCl4 than in cyclohexane. In addition to the structural progression of the solute molecule, the concomitant swelling and shrinking of the solute-solvent cage were also clearly observed. The atom-atom distribution change that appeared above 5 Å is well reproduced by the MD simulation, which indicates the solvation dynamics of newly formed iodine molecules in the course of a vibrational cooling process (Figure 90c,d)
Principal publication and authors
J.H. Lee (a), M. Wulff (b), S. Bratos (c), J. Petersen (d), L. Guerin (b), J.-C. Leicknam (c), M. Cammarata (b), Q. Kong (b), J. Kim (a), K.B. Møller (d) and H. Ihee, (a), J. Am. Chem. Soc. 135, 3255-3261 (2013).
(a) Institute for Basic Science, Center for Time-Resolved Diffraction, Department of Chemistry, KAIST (Republic of Korea)
(b) ESRF
(c) Laboratoire de Physique Théorique de la Matière Condensée, Université Pierre et Marie Curie (France)
(d) Centre for Molecular Movies, Department of Chemistry, Technical University of Denmark (Denmark)
References
[1] R. Jimenez, G.R. Fleming, P.V. Kumar and M. Maroncelli, Nature, 369, 471473 (1994).
[2] W.P. de Boeij, M.S. Pshenichnikov and D.A. Wiersma, Annu. Rev. Phys. Chem. 49, 99123 (1998).
[3] A.L. Harris, J.K. Brown and C.B. Harris, Annu. Rev. Phys. Chem. 39, 341366 (1988).
[4] D.M. Jonas, S.E. Bradforth, S.A. Passino and G.R.J. Fleming, Phys. Chem. 99, 25942608 (1995).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.