- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2010
- Structure of materials
- CO dissociation and transient carbon storage by supported Pd nanoparticles during CO/NO cycling
CO dissociation and transient carbon storage by supported Pd nanoparticles during CO/NO cycling
Heterogeneous catalysts provide the central technology for abating environmental pollution from industrial processes. The preeminent example of this is car exhaust catalysis where the catalyst (Pt/Rh or Pd- nanoparticles dispersed on an oxidic support) has to balance three chemical conversions: the reduction of toxic NOx to benign N2 and the oxidation of hydrocarbons and toxic CO to CO2. A central issue is maintaining high activity levels during the cycling (so-called lambda oscillations) conditions typical of real operation. A basic understanding of how supported noble metal nanoparticles might achieve this is, therefore, of considerable importance.
Beamlines ID24 (dispersive EXAFS) and ID15 (hard X-ray diffraction, HXRD) have collaborated to derive a new way of seeing into the dynamic behaviour of such systems. This approach is an extension of an experiment developed on ID24 to couple time-resolved EXAFS (element specific local structure) to infrared spectroscopy (IR, for surface speciation and molecular functionality) and mass spectrometry (MS) [1]. By using the very hard (89 keV), high flux, X-rays available at ID15B, we can now combine HXRD with IR within the same sample environment developed for time resolved EXAFS/IR. As a result, we have observed previously hidden aspects of how Pd nanoparticles (ca. 3 nm diameter) participate in the oxidation of CO to CO2, how this changes their structure and, subsequently, the disposition of CO molecules adsorbed upon them.
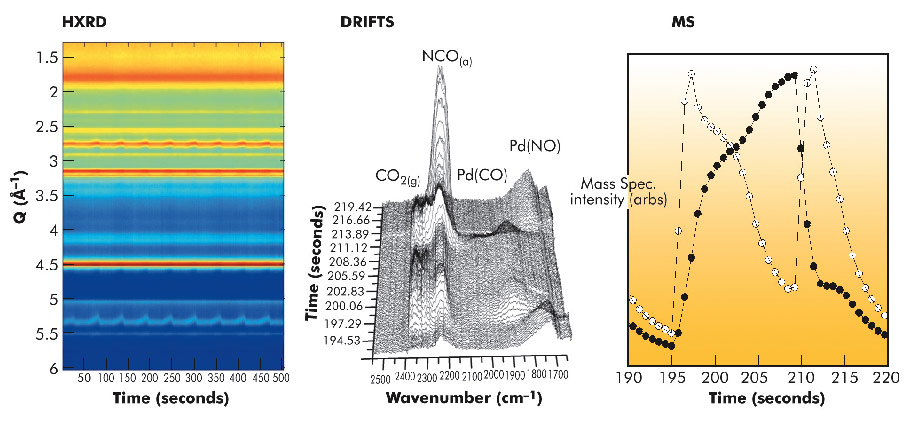
Figure 42 collates the HXRD (for a complete, 10 cycles, CO/NO cycling experiment), IR and MS (for single NO-CO-NO switches) obtained with this experiment. The HXRD shows that reflections due to Pd move as a function of the applied feedstock composition whereas those due to the Al2O3 support do not; that is to say, during the CO cycle the lattice constant of the Pd nanoparticles increases linearly with time. This is rapidly reversed upon switching back to a NO flow.
If we correlate the temporal character of this “breathing” with the MS data we can show that this correlates with a phase of CO2 production from within the CO cycle. The structural change of the nanoparticles revealed by HXRD is directly linked to reactive events that lead to the target conversion of CO to CO2.
These observations yield the following conclusions: during CO exposure an adlayer of molecular CO forms quickly on the Pd nanoparticles. From within that layer a proportion of the CO then dissociates. The atomic oxygen thus released reacts with some of the remaining molecular CO to yield the observed CO2 (MS). The atomic carbon left behind then dissolves into the Pd nanoparticles whose lattice constant increases in proportion to the amount of carbon absorbed. Upon the return to an NO feedstock this stored carbon is then rapidly sequestered to yield CO2 and the lattice constant of the Pd nanoparticles returns to its initial value. This behaviour is not expected on the basis of the chemistry of extended Pd surfaces which do not dissociate CO [2].
 |
|
Fig. 42: DRIFTS, MS, and HXRD from a 2wt% Pd/Al2O3 sample during CO/NO cycling at 673 K. MS closed circles are CO and open circles are CO2. |
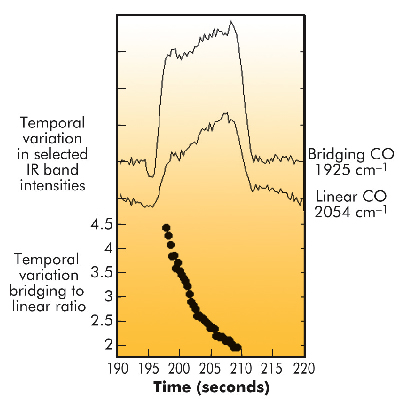
A further important observation arises from correlating the results of the IR spectroscopy with the HXRD. In Figure 43 we show the (IR derived) variation of bridging (Pd2CO) and linear (PdCO) species, as well as their ratio (absorption band intensity: bridged/linear). This shows that the formation of the bulk PdCx as a result of CO dissociation has a secondary effect, changing the partitioning of the remaining molecular CO between sites on the surface of the particles. As carbon is stored within the Pd particle, the occupation of CO in a linearly adsorbed site is significantly promoted.
 |
|
Fig. 43: Temporal variation in IR band intensities due to bridging and linear CO species adsorbed on Pd and their ratio during a single NO:CO:NO cycle at 673 K. |
To summarise we have developed a new experiment for studying working catalysts in a manner that combines IR with HXRD (ID15) or IR/EXAFS (ID24) within the same environment. We have directly observed previously hidden surface and bulk chemistry within a prototypical CO oxidation/NO reduction sequence.
Principal publications and authors
M.A. Newton (a), M. Di Michiel (a), A. Kubacka (b) and M. Fernàndez-Garcià (b), J. Am. Chem. Soc. 132, 4540 (2010); A. Kubacka (b), A. Martínez-Arias (b), M. Fernández-García (b), M. Di Michiel (a) and M.A. Newton (a), J. Catal. 270, 275 (2010).
(a) ESRF
(b) Instituto de Catàlisis y Petroleoquímica, CSIC, C/Marie-Curie 2, Madrid (Spain)
References
[1] M.A. Newton, Topics in Catalysis 52, 1410 (2009).
[2] For instance, R.P.H. Gasser, “An introduction to chemisorption and catalysis by metals” 3rd Ed. OUP (1987).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.