- Home
- News
- Spotlight on Science
- Blue blood: a challenge...
Blue blood: a challenge for structural biologists
23-01-2013
Some of the animals living in the sea have blue blood. The origin of this blue colour is a protein called hemocyanin that contains copper bound to molecular oxygen: an essential interaction allowing these animals to live. Scientists at the ESRF have unravelled the structure of octopus hemocyanin by means of the small-angle X-ray scattering technique in conjunction with data modelling using the algorithm QUAFIT.
A number of invertebrates have blue blood. The blue colour originates from hemocyanin, a copper containing protein that carries molecular oxygen to the tissues instead of the red-coloured protein haemoglobin in the blood of vertebrates. Hemocyanins are giant multimeric molecules: their most distinctive structural features are a tertiary fold and an oligomeric state – features that are recognisable in proteins from various animal groups including octopi, squid, snails, chitons, some bivalves (molluscs), crabs, lobsters, spiders, scorpions, myriapods, and insects (arthropods).
Octopus (Octopus vulgaris) hemocyanin is a truly giant protein with a molecular mass in the order of 3.6 MDa. The protein is a decamer, each monomer having a molecular mass of 385 KDa and comprised of seven functional units (FUs: FUa-FUg) each of ~55 KDa. When the octopus hemocyanin is saturated with oxygen, it carries 70 molecules of oxygen in the blood. The amino acid sequences of the 7 FU’s is already known, and CryoEM studies for hemocyanins have shown how the FUs fit together for other species [1]. For octopus hemocyanin, we set out to investigate its complete quaternary structure, i.e. the association of the FU’s into the monomer and the association of the monomers into hemocyanin, by using synchrotron small-angle X-ray scattering (SAXS) measurements at the ESRF in combination with advanced rigid-body modelling of the structure.
In this study, there were two challenges in particular to be overcome, the reconstruction of the 3-D structure of the complex oligomeric protein and the determination of the various oligomeric aggregate species occurring in a polydisperse system. In fact, this protein represents an ideal SAXS case study because the description of its quaternary structure is centred on the simultaneous modelling of the reciprocal orientation of each FU within the monomer and of the different monomers within the decamer. This point is critical when polydisperse systems are considered, since the polypeptide linkers connecting the FUs give them the freedom to assume different spatial orientations in the dissociated monomers (which could be described as a “string of beads”).
We have addressed these challenges by using a new computational method, QUAFIT [1], which calculates the form factor of an oligomeric protein assembly, finding the optimum space configuration of the monomers. The novelty is that, while fitting the model to the data, the monomer structure is adjusted by searching for the best arrangement of its rigid domains (RDs) as well as for the best conformation of the flexible linkers (FLs) connecting the RDs. In the present case, the seven FUs in the hemocyanin monomer were considered as RDs and their structure was derived by homology modelling from Octopus dofleinii functional unit “g”. (Side bar: Modelling with QUAFIT.)
Small-angle X-ray scattering (SAXS) measurements were carried out at beamline ID02 under buffer conditions either stabilising the decameric form (pH around 7.5 and presence of Ca2+ ions, conditions that mimic the in vivo situation) or dissociating the oligomer into subunits of different aggregation states (alkaline pH, absence of Ca2+ ions). Our various solution conditions, although different to the conditions in octopus hemolymph, were chosen to challenge the quaternary state of the protein and produce many of the possible oligomers by dissociation. The decamers are stable over a wide range of experimental conditions provided that the pH is around neutrality and Ca2+ ions are present in solution, a situation encountered in vivo by octopus hemolymph. The SAXS patterns of Octopus vulgaris hemocyanin both in decameric and monomeric forms are reported in Figure 1, together with their structures derived by QUAFIT. The structure of the decamer (Figure 1b) is in very good agreement with recently published cryo-electron microscopy (cryo-EM) images [2], while the structure of the dissociated “loose” monomer is found to be well represented by a distribution of many conformations (Figure 1a).
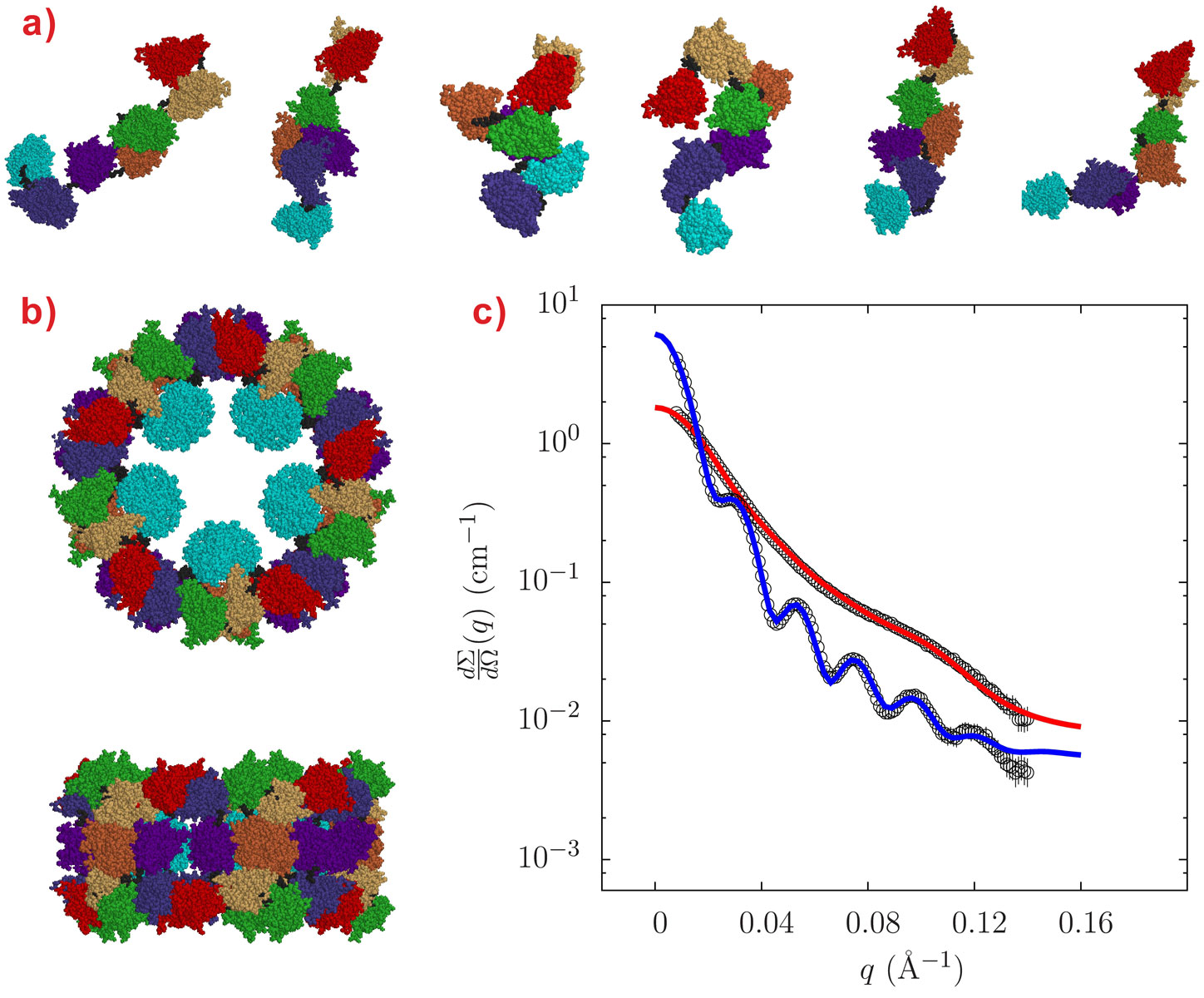 |
|
Figure 1. Gallery of structures of Octopus vulgaris hemocyanin as determined by QUAFIT analysis of SAXS data: a) Monomeric states; b) Decameric state, axial and lateral views. The different functional units (or rigid domains RDs as defined in QUAFIT) are: RD-a, red; RD-b, yellow; RD-c, green; RD-d, orange; RD-e, purple; RD-f, blue; RD-g, cyan; the polypeptide stretches that connect them (flexible linkers as defined in QUAFIT) are in black. d) The plot shows the results of QUAFIT fitting of SAXS data for decameric hemocyanin (blue line) or monomeric hemocyanin (red line). |
The SAXS data also confirm that the aggregation state of octopus hemocyanin strongly depends on the buffer conditions (Figure 2). While the presence of different intermediate oligomers does not hamper the structural reconstruction by cryo-EM since image reconstruction methods allow each individual particle to be singled out in the microscope field, the polydispersity may represent a serious problem in SAXS analysis because the experimental patterns depend on the form factor of all the oligomers in solution. However, hierarchical series of intermediate oligomeric species can be described by the QUAFIT method on the basis of the best monomer structures. In Figure 2, fitting curves are superposed on SAXS patterns obtained under a variety of experimental conditions: the modelling results are reported in terms of histograms presenting the molar fraction of the different aggregates and of the hierarchical structures built up by the assembly of the “compact” monomer.
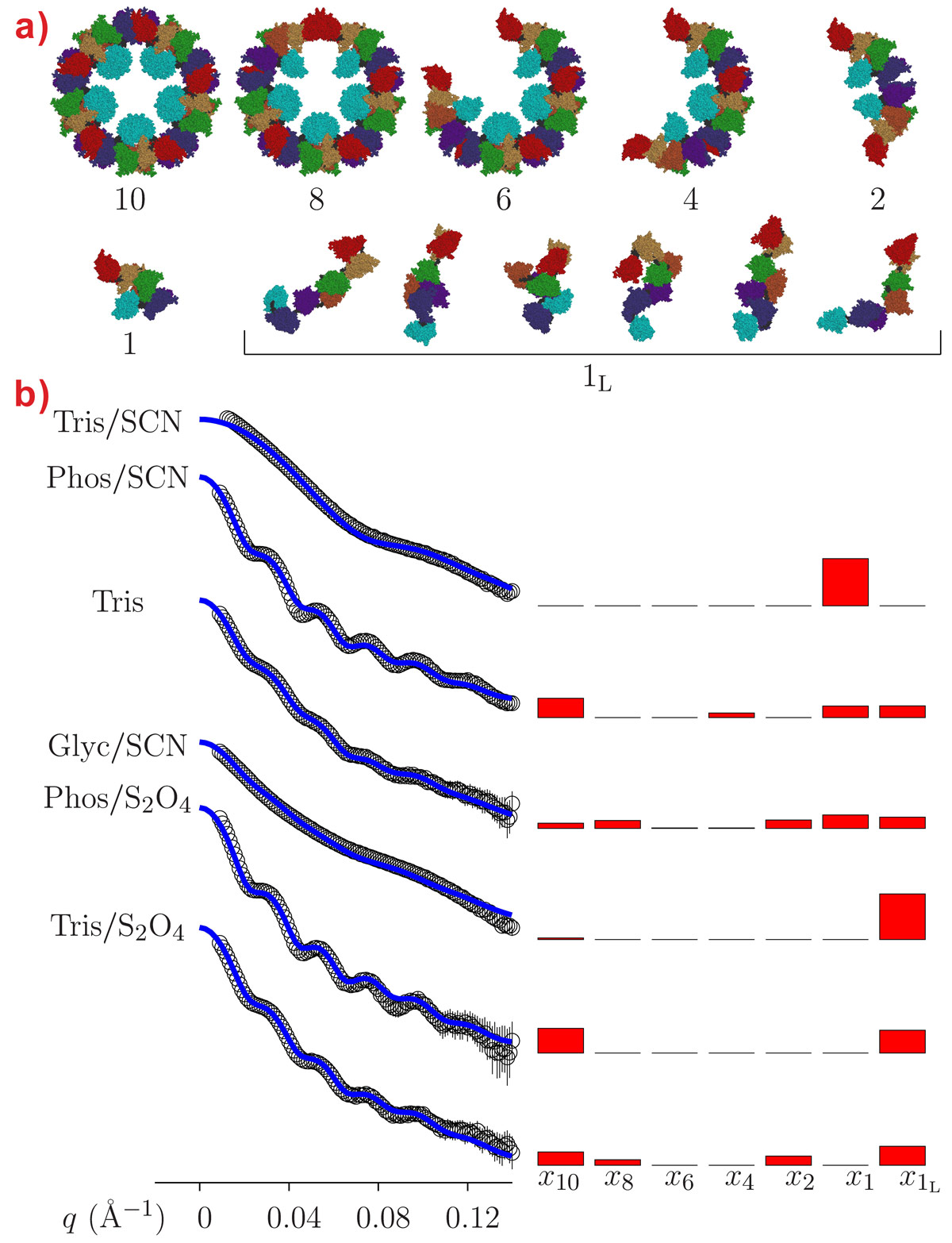 |
|
Figure 2. a) Gallery of quaternary structures of Octopus vulgaris hemocyanin found in various buffer conditions. Monomeric hemocyanin exists as “loose” monomer (1L) or as “compact” or aggregation-prone monomer (1). By hierarchical aggregation, the “compact” monomer produces dimers (2), tetramers (4), hexamers (6), octamers (8) and decamers (10). b) SAXS data and histograms showing the molar fraction of each oligomer for various buffer conditions. The molar fraction was determined by QUAFIT analysis (blue lines) of SAXS data; x1L representing the molar fraction of the “loose” monomer, x1 of the “compact monomer” xn - x10 of the different intermediate oligomers. |
Structural biologists are increasingly interested in the study of complex oligomeric aggregates since a protein usually functions as a macromolecular complex rather than as a single molecule. Furthermore, the rapid development of proteomics requires methods that allow the structure of biological machineries to be determined from the knowledge of their main building blocks and that also enable the description of structural hierarchies which can be simultaneously present in the system. The combination of SAXS and data modelling by QUAFIT has provided a detailed view of the structure of hemocyanin, for both the native oligomer and its constituent monomer. These tools should be useful to aid interpretation of many other complex protein structures.
Principal publication and authors
F. Spinozzi (a), P. Mariani (a), I. Mičetić (b,c), C. Ferrero (d), D. Pontoni (d), M. Beltramini (b), PLoS One 11, e49644 (2012).
(a) Department DiSVA, Marche Polytechnic University and CNISM, Ancona (Italy)
(b) Department of Biology, University of Padova, (Italy)
(c) present address: ECSIN-European Center for the Sustainable Impact of Nanotechnology, Veneto Nanotech S.C.p.A., Rovigo (Italy)
(d) ESRF
References
[1] C. Gatsogiannis, A. Moeller, F. Depoix, U. Meissner, J. Markl, J. Mol. Biol. 374, 465 (2007).
[2] F. Spinozzi and M. Beltramini, Biophys. J. 103, 511 (2012).
Modelling with QUAFIT
The main functions of the QUAFIT algorithm [2] are summarised in the following:
- The structure of the protein assembly, which can follow the symmetry laws of a proper point group and/or a screw axis, is determined through a hierarchical series of intermediate oligomeric species, formed by the optimum arrangement of an “asymmetric unit” (the monomer).
- Many hierarchical series, corresponding to different configurations of the asymmetric monomer and/or different symmetries of the assembly, may simultaneously exist in the buffer solution, and the molar fraction of each component of any series is determined by the SAS data fitting.
- The asymmetric monomer is described by the best arrangement (position and orientation) of the RDs and by the fittest conformation (in terms of Ramachandran and side chain angles for each residue) of the FLs connecting the RDs. Depending on the hierarchical series, the monomer can exist in one configuration or as a mixture of different configurations.
- By means of the spherical tensors algebra, the small-angle scattering amplitude of the entire assembly (and/or of each intermediate oligomer) is expressed as a linear combination of the partial amplitudes of RDs and FLs (calculated over an atomic basis), previously symmetrised with respect to the assembly symmetry.
- The control of the compactness of the assembly is obtained in two steps: by calculating the “contact distances” between RDs as a function of their mutual orientation through a series expansion of Stone’s rotational invariants by introducing in the merit function to be minimised an asymmetric Lennard-Jones potential based on the “contact distances” found in the first step.
- The control of the capability of each FL to properly connect two RDs is also done in two steps: - first - the chain is left “free” to grow from one of the two RDs and, for each tentative conformation, the average distance between the positions of four atoms on the free end of the FL chain is calculated with respect to the positions that they should occupy in the other RD, and – second - an elastic potential term based on the average distance found in the first step is introduced into the merit function.
- The merit function, which includes the standard χ2 related to the SAXS curve, the Lennard-Jones potential and the elastic potential introduced in 5) and 6), respectively, is minimised by a simulated annealing protocol.
Top image: Hemocyanin is a giant decamer. By changing the solvent conditions it dissociates into the monomers formed from seven rigid units connected by six flexible linkers. SAXS curves for hemocyanin and a monomer mixture are shown in blue and red.
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.